
Reactivity descriptor for the retro Diels–Alder reaction of partially saturated 2-pyrones: DFT study on substituents and solvent effects†
Abstract
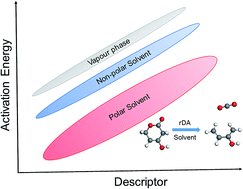
The retro-Diels–Alder (rDA) reaction of partially saturated 2-pyrone molecules to form the 1,3-butadiene backbone and CO2 was studied using density functional theory (DFT) calculations in vapor-phase, polar and non-polar solvents. The activation barriers for the ring-opening and decarboxylation of the molecules were correlated to the type of substituent present on the 2-pyrone ring. In the vapor-phase, the electronic effect of substituents led to a linear scaling relationship between the calculated activation barrier and corresponding frontier molecular orbital (FMO) gap of the product diene and CO2. A new descriptor was proposed as the average of the ionization potential (IP) of the diene and the electron affinity (EA) of the dienophile to describe the activation energy trend. Solvents were calculated to reduce the activation barriers by stabilizing the polar transition state by as much as 40 kJ mol−1, wherein polar solvents were calculated to reduce the barrier more than the non-polar solvents. The rDA reaction activation barrier in the solvent decreases in the following order: vapor-phase > n-hexane > benzene > acetone > methanol > water. The effect of solvents in rDA reactivity trends was successfully described for the first time through a single descriptor, the FMO gap. The existence of a Brønsted–Evans–Polanyi (BEP) relationship was established for the rDA reaction over different solvents. In solvents, the FMO gap, (IPdiene + EAdienophile)/2 and BEP relationship were proposed as the reactivity descriptors for the rDA reaction of 2-pyrones.
 Please wait while we load your content...
Please wait while we load your content...

