
Electrically conductive nanofibrous scaffolds based on poly(ethylene glycol)s-modified polyaniline and poly(ε-caprolactone) for tissue engineering applications
Maryam Hatamzadehab,
Peyman Najafi-Moghadama,
Younes Beygi-Khosrowshahicd,
Bakhshali Massoumi*b and
Mehdi Jaymand*e
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Urmia, P. O. Box: 57561-51818, Urmia, Iran
bDepartment of Chemistry, Payame Noor University, P. O. Box: 19395-3697, Tehran, Iran. E-mail: b_massoumi@pnu.ac.ir; bakhshalim@yahoo.com
cStem Cell and Tissue Engineering Research Laboratory, Sahand University of Technology, P.O. Box: 51335-1996, Tabriz, Iran
dChemical Engineering Department, Faculty of Engineering, Azarbaijan Shahid Madani University, P.O. Box: 53751-71379, Tabriz, Iran
eResearch Center for Pharmaceutical Nanotechnology, Tabriz University of Medical Sciences, P.O. Box: 51656-65811, Tabriz, Iran. E-mail: m_jaymand@yahoo.com; m.jaymand@gmail.com; jaymandm@tbzmed.ac.ir; Fax: +98-41-33367929; Tel: +98-41-33367914
First published on 28th October 2016
Abstract
The purpose of this study was to design and develop electrically conductive nanofibrous scaffolds based on poly(ethylene glycol)s-modified polyaniline [PEGs-b-(PANI)4] and poly(ε-caprolactone) (PCL) for tissue engineering applications. For this purpose, two AB4 miktoarm star-shaped conductive PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4 were synthesized through a multistep process started from diepoxylated PEGs, and subsequently hydrolyzed to PEGs ends-caped tetraol [PEGs(OH)4]. Afterward, phenylamine-functionalized PEGs AB4 macromonomers (PhAPEGsM) were synthesized by functionalization of PEGs(OH)4 with p-anthranilic acid. The macromonomers obtained were subsequently used in chemical oxidation copolymerization with aniline monomer to produce AB4 miktoarm H-shaped conductive polymers. The solutions of the synthesized polymers and PCL were electrospun to produce uniform, conductive, and biocompatible nanofibers. Some physicochemical properties of these nanofibers such as morphologies, electrical conductivities, hydrophilicities, and mechanical properties were investigated. The biocompatibilities of the fabricated nanofibers were confirmed by assessing the adhesion, viability and proliferation of mouse fibroblast L929 cells using SEM and MTT assay, respectively. As the results, of the conductivities, biocompatibilities, hydrophilicities, and mechanical properties assessments it is demonstrated that these nanofibers are potentially suitable as scaffolds for use in tissue engineering that requires electroactivity.
1. Introduction
Nowadays, tissue and organ failure resulting from diseases, injury or other types of damages is one of the most important health problems with an increasing incidence worldwide. Tissue engineering (TE) is a relatively novel interdisciplinary field that introduced for repair or regeneration of failed tissues/organs using living cells, scaffolds and signal molecules as the three key fundamental elements.1–5 Among these, scaffolds as artificial extra cellular matrices (ECMs) have a pivotal role in TE quality, in part due to their influence on cell attachment, orientation, proliferation, migration, differentiation, and final neo-tissue formation.6–8 The fundamental requirements for a proper scaffold can be listed as:(1) Adequate biocompatibility and biodegradability.
(2) Suitable structures and mechanical characteristics.
(3) Proper surface topography and chemical composition.
(4) Simple and cost effective fabrication technology.9,10
Some methodologies such as gas foaming,11 freeze drying,12 phase separation,13 solvent casting/particle leaching,14 particle pressing,15 weaving/knitting,16 rapid prototyping (RP),17 photolithography,18 and electrospinning19 have been developed or introduced for scaffolding. Among these, the electrospinning is a powerful technique to prepare nanofibrous scaffolds to mimic the architecture and biological functions of the ECM.19–21 This scaffolding technique has stimulated a great deal of interest due to its some advantages such as similar morphology to the human native ECM, ultra-thin continuous nanofibers (ranging from 5 to 500 nm), tunable pore size distribution, high porosity with high surface area, applicability for both organic and inorganic materials, simplicity, scalability, and more cost-effectivity.19,22
On the other hand, it is well accepted that each cell produces a membrane potential in the range of few to tens mV, that influence most functionality of the cells such as polarity, embryogenesis, wound healing, differentiation, division, and motility.23,24 From this point of view, applying electrical stimulation (ES) through the scaffold can be regulates cell functions especially for electrically excitable cells such as myoblasts, fibroblasts, osteoblasts, chick embryo dorsal root ganglia, and neural crest cells.25,26 Therefore, the fabrication of electrically conductive scaffolds is a challenging step for a successful regeneration of tissues/organs (e.g., skin, nerve, bone, and muscle) that require electroactivity. In this respect, some researcher proposed the incorporation of conductive nanosystems (NSs) such as gold nanowires,27 and carbon-based materials (e.g., carbon nanotube, and graphene)28 into implantable polymeric scaffolds to induce electrical conductivity for modulate cellular behavior. However, the main drawbacks of these systems are the non-biodegradability and in vivo long-term effects of applied fillers.21,29 From the practical point of view, the intrinsically conductive polymers (ICPs) such as polyaniline (PANI), polypyrrole (PPy), polythiophene (PTh), and their derivatives can be considered as potential candidates for overcoming the above mentioned problems.21,29,30 The development of ICPs-based materials for biomedical applications expanded greatly from the 1980s when it was found that these polymers were compatible with many cells/tissues in their purified forms both in vitro and in vivo, which are further enhanced by blending or covalently grafting with other synthetic, semi-synthetic, and natural biodegradable and biocompatible polymers.31,32 Among these, the electrically conductive scaffolds based on PANI has received more interest due to its antioxidant property which can act as free radical scavenger at the site of injury, and minimizing scar formation.33,34
The objective of this work was to synthesis and characterization of AB4 miktoarm star-shaped conductive poly(ethylene glycol)s-modified polyaniline [PEGs-b-(PANI)4], and preparation of its electrospun nanofibers with poly(ε-caprolactone) (PCL) as nanofibrous scaffolds for tissue engineering applications. For this purpose, the diepoxylated PEGs (Mn = 2000 and 6000 g mol−1) were hydrolyzed to PEGs ends-caped tetraol and then functionalized with p-anthranilic acid to afford phenylamine-functionalized PEGs AB4 macromonomers (PhAPEG2000M and PhAPEG6000M). The macromonomers obtained were subsequently used in chemical oxidation copolymerization with aniline monomer to afford AB4 miktoarm H-shaped conductive polymers. The solutions of synthesized polymers and PCL were electrospun to produce uniform, conductive, and biocompatible nanofibers. The biocompatibilities of the nanofibers were evaluated through adhesion, viability and proliferation of the mouse fibroblast L929 cell line using SEM and MTT assay, respectively.
2. Experimental
2.1. Materials
Poly(ε-caprolactone) (Mn = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–90000 g mol−1), poly(ethylene glycol) (Mn = 2000 and 6000 g mol−1), camphorsulfonic acid (CSA), sodium hydroxide (NaOH), p-toluene sulfonic acid (p-TSA), and p-anthranilic acid were purchased from Sigma-Aldrich (USA) and were used as received. Aniline monomer was purchased from Merck (Darmstadt, Germany), and was distilled twice under reduced pressure before use. Epichlorohydrin (Merck) was dried by calcium hydride (CaH2), and distilled twice under reduced pressure before use. Ammonium peroxydisulfate (APS; Merck) was purified through re-crystallization from ethanol at room temperature. Toluene, xylene, and tetrahydrofuran (THF) were purchased from Merck, dried by refluxing over sodium, and distilled under argon prior to use. All other reagents were purchased from Merck or Sigma-Aldrich and purified according to the standard methods.
000–90000 g mol−1), poly(ethylene glycol) (Mn = 2000 and 6000 g mol−1), camphorsulfonic acid (CSA), sodium hydroxide (NaOH), p-toluene sulfonic acid (p-TSA), and p-anthranilic acid were purchased from Sigma-Aldrich (USA) and were used as received. Aniline monomer was purchased from Merck (Darmstadt, Germany), and was distilled twice under reduced pressure before use. Epichlorohydrin (Merck) was dried by calcium hydride (CaH2), and distilled twice under reduced pressure before use. Ammonium peroxydisulfate (APS; Merck) was purified through re-crystallization from ethanol at room temperature. Toluene, xylene, and tetrahydrofuran (THF) were purchased from Merck, dried by refluxing over sodium, and distilled under argon prior to use. All other reagents were purchased from Merck or Sigma-Aldrich and purified according to the standard methods.
2.2. Synthesis of α,ω-diepoxy-PEGs
A 250 mL dried three-necked round-bottom flask equipped with condenser, septum, gas inlet/outlet, and a magnetic stirrer was charged with PEG2000 (20.0 g; 10 mmol), and dried toluene (100 mL). The flask was joined to the argon gas line, and the reaction mixture was de-aerated by bubbling highly pure argon for 10 minutes. Afterward, NaOH (1.0 g; 25 mmol) was added to the flask under argon protection. The reaction mixture was stirred for about 1 hour at room temperature, and then dried epichlorohydrin (2.52 mL; 30 mmol) was introduced with a syringe through the septum. The flask placed in a silicon oil bath at 50 ± 3 °C and refluxed for about 7 hour under an inert atmosphere. At the end of this time, the content of the flask was poured into a large amount of cold diethyl ether. The product was filtered, washed with diethyl ether several times, and dried in reduced pressure at room temperature to afford α,ω-diepoxy PEG2000.The α,ω-diepoxy PEG6000 was synthesized by the same procedure with the appropriate amounts of PEG6000 (30.0 g, 5 mmol), dried toluene (150 mL), NaOH (0.50 g; 13 mmol), and epichlorohydrin (1.25 mL; 15 mmol).
2.3. Synthesis of PEGs ends-caped tetraol [PEGs(OH)4]
A 100 mL round-bottom flask equipped with a condenser, and a magnetic stirrer, was charged with α,ω-diepoxy PEG2000 (10 g, 5 mmol), and NaOH solution (70 mL; 1 mol L−1). The reaction mixture was stirred for about 24 hours at 50 ± 3 °C. At the end of this time, the mixture was frozen and lyophilized. The crude product was purified by dialyzing in distilled water (one liter) using 1000 molecular weight cut-off dialyzed bag, in order to remove residual NaOH. The product was filtered, and dried using a freeze drying system to afford PEG2000(OH)4. The PEG6000(OH)4 was synthesized by the same procedure.2.4. Synthesis of phenylamine-functionalized PEGs AB4 macromonomers (PhAPEGsM)
A 100 mL three-neck round-bottom flask equipped with a dean-stark trap, gas inlet/outlet, and a magnetic stirrer, was charged with PEG2000(OH)4 (4 g, 2 mmol), p-anthranilic acid (1.37 g, 10 mmol), and anhydrous xylene (80 mL). A catalytic amount of p-TSA (0.10 g, 0.56 mmol) as the dehydrating agent was added to the flask, and the mixture was de-aerated by bubbling highly pure argon for 15 minutes. The flask placed in a silicon oil bath at 140 ± 3 °C and refluxed for about 6 hours. The water formed during the reaction was removed as an azeotrope until no more water was formed, which could indicate that the reaction had gone to completion. At the end of this time, the flask was rapidly cooled to room temperature by ice/water bath. The crude product was precipitated by adding cold diethyl ether, and then extracted with dichloromethane for three times, in order to remove un-reacted p-anthranilic acid. The polymer solution was precipitated into large amount of cold diethyl ether, and dried in reduced pressure at room temperature. It should be pointed out that the p-anthranilic acid is not soluble in dichloromethane, while PhAPEG2000M is soluble in this solvent. The PhAPEG6000M was synthesized by the same procedure.2.5. Synthesis of H-shaped miktoarm PEGs-b-(PANI)4
The AB4 miktoarm H-shaped PEG2000-b-(PANI)4 was synthesized by an interfacial polymerization method as follows. In a 250 mL beaker APS (1.37 g, 6 mmol) was dissolved in 80 mL of sulfuric acid solution (1 mol L−1). In a separate container, PhAPEG2000M (0.5 g, 0.25 mmol), and aniline monomer (1.0 g, 10.7 mmol) were dissolved in chloroform (80 mL). This mixture was added gently and with minimal agitation along the sides of the above mentioned beaker. The aniline/PhAPEG2000M/chloroform solution form the lower organic layer and APS solution forms the upper aqueous layer. An overnight reaction time is generally sufficient. Then, the solid polymer that consists of homo-PANI and AB4 miktoarm H-shaped PEG2000-b-(PANI)4 was filtered, washed with methanol several times (in order to remove unreacted PhAPEG2000M chains, oligomers, and by-products), and dried in vacuum at room temperature.The crude product was extracted whit THF in a Soxhlet apparatus for 24 hours, in order to remove any homo-PANI chains. According to our tests, the synthesized PEG2000-b-(PANI)4 is soluble in THF, while homo-PANI is not soluble in THF. The polymer solution was filtered, concentrated by a rotary evaporator, precipitated into excess cold methanol, and dried in reduced pressure at room temperature. The AB4 miktoarm H-shaped PEG6000-b-(PANI)4 was synthesized by the same procedure.
2.6. Synthesis of CSA-doped PEGs-b-(PANI)4
The sulfuric acid-doped PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4 (emeraldine salts) were later converted to their base form by stirring for about 5 hours in ammonia solution (100 mL; 5% v/v). The un-doped polymers were filtered, and washed several times with excess water followed by methanol. The obtained dark blue powders were dried under vacuum at room temperature. Afterward, the polymers were added to aqueous solutions of CSA (2 mol L−1), and stirred for about 7 hours to afford CSA-doped PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4. At the end of this time, the polymers were filtered, washed several times with excess water followed by methanol, and dried in vacuum at room temperature.2.7. Fabrication of PEGs-b-(PANI)4/PCL electrospun nanofibers
The PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL nanofibers were prepared by electrospinning of the same volume solutions of PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4 (in dimethylsulfoxide; DMSO; 2% w/v), and PCL (chloroform:2-chloroethanol 2:1 (v/v); 3% w/v) as described in our previous works.22,35
2.8. Biocompatibility analysis
:1 v/v), and after then the clear blue viable cells were count using hemocytometer slide.2.9. Characterization
Fourier transform infrared (FTIR) spectra of the synthesized samples were collected on a Shimadzu 8400S FTIR (Shimadzu, Kyoto, Japan) in the range of 4000 to 400 cm−1 with a resolution of 4 cm−1 using potassium bromide (KBr) pellet technique. Proton nuclear magnetic resonance (1H NMR) spectra of the samples were recorded at 25 °C using an FT-NMR (400 MHz) Bruker spectrometer (Bruker, Ettlingen, Germany). The sample for NMR spectroscopy was prepared by dissolving about 10 mg of samples in 1 mL of deuterated chloroform (CDCl3) or dimethyl sulfoxide (DMSO-d6), and chemical shifts were reported in ppm units with tetramethylsilane (TMS) as an internal reference. The scanning electron microscope (SEM) type 1430 VP (LEO Electron Microscopy Ltd, Cambridge, UK) was applied to determine the surface morphologies of the synthesized samples. Ultraviolet-visible (UV-vis) spectra were recorded with a Shimadzu 1650 PC UV-vis spectrophotometer (Shimadzu, Kyoto, Japan). Electrochemical experiments were performed using an Auto-Lab PGSTA T302N electrochemical analysis system and GPES 4.7 software package (ECO chemie, The Netherlands). The electrochemical cell contained five openings: three of them were used for the electrodes (working, counter, and reference), and two for argon bubbling in the solutions during all experiments. The conductivities of the samples were determined using the standard four-probe technique (Azar Electrode, Urmia, Iran) at room temperature. The thermal stability of the synthesized samples were investigated by means of thermogravimetric analyzer (TGA-PL STA 1640 equipment (Polymer Laboratories, Shropshire, UK)). The thermogravimetric analysis (TGA) experiment was conducted under nitrogen atmosphere from room temperature to 650 °C with heating rate of 10 °C min−1. The ultimate tensile strength and strain to break were determined using a Zwick tensile testing machine (Z010, Zwick/Roell, Ulm, Germany). The wettabilities of the fabricated nanofibers were investigated by water drop contact angle measurement using an OCA 20 plus contact angle meter system (Data Physics Instruments GmbH, Filderstadt, Germany). The droplet size was 5 μL, and at least five samples were used for each test.3. Results and discussion
The emergence of nanotechnology has provided new possibilities for development of novel biomaterials for biomedical applications. Nanofibrous polymeric scaffolds for regenerative medicine applications are one of the recent advances in the field of nanotechnology, which led to new opportunities for mimicking of native ECM as a key requirement for an ideal artificial scaffold. Nevertheless, this field is still growing and many issues remain to be addressed. The overall methodologies for the synthesis of H-shaped miktoarm PEGs-b-(PANI)4 and fabrication of nanofibrous electrically conductive scaffolds for tissue engineering applications are shown in Schemes 1 and 2.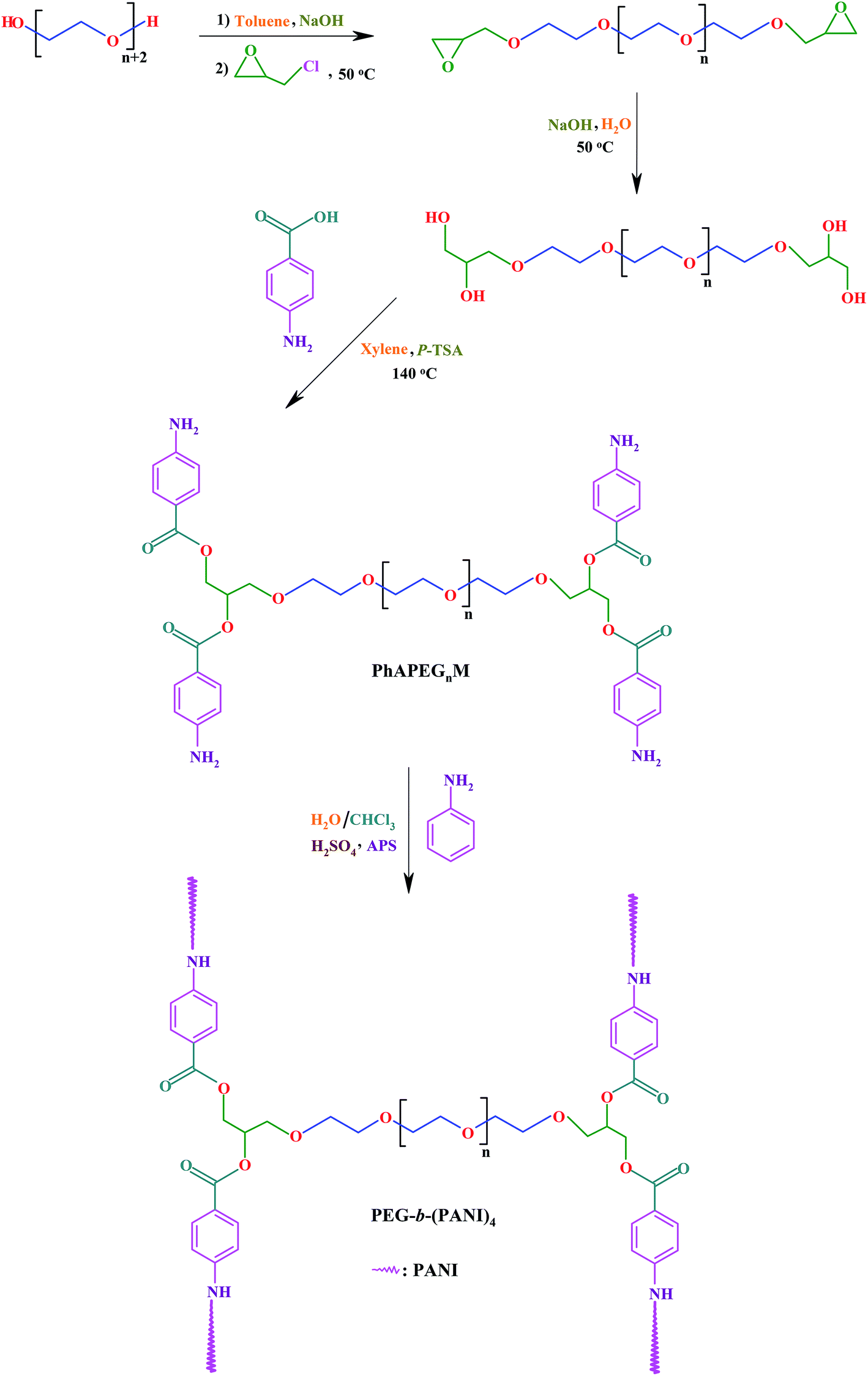 | ||
| Scheme 1 The overall methodologies for synthesis of H-shaped miktoarm PEGs-b-(PANI)4. | ||
 | ||
| Scheme 2 The overall strategy for the fabrication of nanofibrous biocompatible, porous, and electrically conductive scaffolds based on PEGs-b-(PANI)4 and PCL for tissue engineering applications. | ||
3.1. Synthesis of α,ω-diepoxy PEGs
The chemical structures of α,ω-diepoxy PEGs were characterized by means of FTIR and 1H NMR spectroscopies as shown in Fig. 1 and 2. The FTIR spectrum of the pure PEG2000 (Fig. 1a) shows the characterization absorption bands due to the stretching vibrations of aliphatic C–H at 2950–2800 cm−1 region, C–H bending vibrations at 1466 and 1358 cm−1, C–O stretching vibration at 1109 cm−1, and hydroxyl stretching vibration as a broad band centered at 3488 cm−1. The FTIR spectrum of the PEG6000 showed similar bands (the spectrum is not shown here). After incorporation of epoxy group into the PEG2000 and PEG6000 ends, the most significant change in the FTIR spectra is the disappearance of stretching vibration of hydroxyl groups at 3488 cm−1. This verifies that most of the hydroxyl groups were converted to epoxide groups (Fig. 1b and c). | ||
| Fig. 1 The FTIR spectra of pure PEG2000 (a), α,ω-diepoxy PEG2000 (b), and α,ω-diepoxy PEG6000 (c). | ||
 | ||
| Fig. 2 The 1H NMR spectra of pure PEG2000, α,ω-diepoxy PEG2000, and α,ω-diepoxy PEG6000. | ||
The synthesis of α,ω-diepoxy PEGs were further verified by 1H NMR spectroscopy as shown in Fig. 2. The 1H NMR spectrum of the pure PEG2000 showed the chemical shifts at 3.30–3.90 ppm related to the O–CH2 groups of PEG backbone (a and b), and hydroxyl end groups at 2.30–2.70 ppm (c). In 1H NMR spectra of the α,ω-diepoxy PEG2000 and α,ω-diepoxy PEG6000 samples the appearances of chemical shifts at 3.00–3.20 (d), 2.65–2.80 (f), and 2.40–2.60 (e) ppm are verified the successful incorporation of epoxy groups into the PEG2000 and PEG6000 ends. It should be pointed out that, as seen in 1H NMR spectra of the α,ω-diepoxy PEGs the chemical shifts related to the hydroxyl end groups at 2.30–2.70 ppm is completely disappeared. This verifies that approximately all of the hydroxyl ends groups were converted to epoxide groups, in accordance with the results obtained by FTIR spectroscopy.
3.2. Synthesis of PhAPEGsM macromonomers
The PhAPEGsM macromonomers were synthesized through the esterification of PEGs ends-caped tetraol [PEGs(OH)4] with p-anthranilic acid in the presence of p-TSA as the catalyst. In recent years, p-TSA considered as an efficient catalyst in different areas of organic synthesis, mainly due to its convenient reaction process, cost effectiveness, recoverability, the excellent functional group tolerance, and environmentally friendly. In the case of esterification reactions, it is well demonstrated that, p-TSA can be used as an effective catalyst.36,37The FTIR spectra of the PEG2000(OH)4 (a), PEG6000(OH)4 (b), PhAPEG2000M (c), and PhAPEG6000M (d) are shown in Fig. 3. As seen in this figure, after basic hydrolysis of α,ω-diepoxy PEGs to PEGs(OH)4 the most distinctive feature in the FTIR spectra are the appearance of broad absorption band centered at about 3450 cm−1 related to four hydroxyl ends groups in each sample. The successful synthesis of PhAPEGsM macromonomers are verified by the appearances of some new bands as follows. The stretching vibrations of carbonyl groups at 1692 cm−1, the stretching vibrations of amine groups as a broad band centered at 3450 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations at about 1600 cm−1, and γ(C–H) in the aromatic ring at 698 cm−1.
C stretching vibrations at about 1600 cm−1, and γ(C–H) in the aromatic ring at 698 cm−1.
 | ||
| Fig. 3 The FTIR spectra of the PEG2000(OH)4 (a), PEG6000(OH)4 (b), PhAPEG2000M (c), and PhAPEG6000M (d). | ||
The synthesized PEGs(OH)4 and PhAPEGsM macromonomers are further characterized by means of 1H NMR spectroscopy (Fig. 4). As seen in 1H NMR spectrum of the PEG2000(OH)4 after basic hydrolysis of α,ω-diepoxy PEGs to PEGs(OH)4 the chemical shifts related to epoxy groups at 2.40–3.20 ppm are completely disappeared and the chemical shift corresponding to the hydroxyl groups is appeared at 2.30–2.70 ppm. The 1H NMR spectrum of the PEG6000(OH)4 showed similar chemical shifts with minor differences (the spectrum is not shown here).
 | ||
| Fig. 4 The 1H NMR spectra of the PEG2000(OH)4 and PhAPEG2000M. | ||
The successful synthesis of PhAPEGsM macromonomers were further approved by 1H NMR spectroscopy. As seen in 1H NMR spectrum of the PhAPEG2000M the incorporation of phenylamine groups into the PEGs ends is verified by the appearances of new chemical shifts at 6.55–6.75 and 7.85–7.95 ppm related to the aromatic protons of the phenylamine groups. It should be noted that the chemical shifts at 2.45 ppm related to hydroxyl group showed some of them dose not reacted with p-anthranilic acid. The 1H NMR spectrum of the PhAPEG6000M showed similar chemical shifts with minor differences (the spectrum is not shown here). The 1H NMR data revealed that the degree of conversion were 92 and 86% by mol for PhAPEG2000M and PhAPEG6000M, respectively.
3.3. Characterization of H-shaped miktoarm PEGs-b-(PANI)4
C at 1596 and 1475 cm−1, Caromatic–N stretching vibration at 1301 cm−1, γ(C–H) in the aromatic ring at 807 and 691 cm−1, and the stretching vibration of SO group related to dopant (CSA) as a strong band at 1137 cm−1.
 | ||
| Fig. 5 The FTIR spectra of the PEG2000-b-(PANI)4 (a), PEG6000-b-(PANI)4 (b), and pure PANI (c). | ||
The FTIR spectra of the PEG2000-b-(PANI)4 (Fig. 5a), and PEG6000-b-(PANI)4 (Fig. 5b) shows the all characteristic absorption bands related to both PEG and PANI segments. In comparison with the FTIR spectrum of the pure PANI the most significant changes in the both FTIR spectra of the PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4 are the appearances of C–H stretching vibrations at 3100–2800 cm−1 region.
The synthesized PEG2000-b-(PANI)4 (Fig. 6a), and PEG6000-b-(PANI)4 (Fig. 6b) were further characterized by means of 1H NMR spectroscopy as shown in Fig. 6. As seen in this figure, the growth of PANI segments from PhAPEG2000M and PhAPEG6000M were verified by the appearance of new chemical shifts at 7.30–7.65 ppm related to the aromatic protons of PANI segments. Other chemical shifts are labeled in the 1H NMR spectrum of the PEG6000-b-(PANI)4 sample. These FTIR and 1H NMR spectra assignments verify that the aniline has grown onto PhAPEG2000M and PhAPEG6000M, and then the PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4 were successfully synthesized.
 | ||
| Fig. 6 The 1H NMR spectra of the PEG2000-b-(PANI)4 (a), and PEG6000-b-(PANI)4 (b). | ||
 | ||
| Fig. 7 The SEM images of the PEG2000-b-(PANI)4 (top) and PEG6000-b-(PANI)4 (bottom) samples at different magnifications. | ||
 | ||
| Fig. 8 Thermogravimetric analysis (TGA) of the pure PEG2000 (a), PEG6000-b-(PANI)4 (b), PEG2000-b-(PANI)4 (c), and pure PANI (d) samples. | ||
The main decomposition of the PEG6000-b-(PANI)4 (Fig. 8b) was also occurring in one step around 230–400 °C, and after which the loss rate slows down. The residue at 650 °C for this sample is 14 wt%. In similar, the PEG2000-b-(PANI)4 sample (Fig. 8c) exhibits a one-step decomposition process at around 240–470 °C, and after which the loss rate slows down. The residue at 650 °C for this sample is 19 wt%. The initial weight loss between 120–230 °C for both PEG6000-b-(PANI)4 (∼11 wt%) and PEG2000-b-(PANI)4 (∼8 wt%) samples may be related to the evaporation of solvent or moisture, and degradation of small molecules. According to results obtained from TGA, the conclusion could be drawn that the grafting of PANI onto functionalized PEGs enhanced their thermal stabilities.
 | ||
| Fig. 9 Cyclic voltammetry curves (CVs) of the synthesized pure PANI (a), PEG2000-b-(PANI)4 (b), and PEG6000-b-(PANI)4 (c) samples, at a scan rate of 10 to 50 mV s−1, in an aqueous solution of CSA (1.0 mol L−1) between −0.60 to +1.20 V, and the linear relationship between the current and scan rate in the mentioned synthesized samples (d) (the currents in the samples are related to the first anodic peaks). | ||
In similar, the CVs of PEG2000-b-(PANI)4 sample (Fig. 9b) shows two typical redox couples with anodic peaks at approximately 0.40 and 0.62 V versus the Ag/AgCl electrode. The CVs of the PEG6000-b-(PANI)4 sample (Fig. 9c) also shows two typical redox couples with anodic peaks at approximately 0.42 and 0.63 V versus the Ag/AgCl electrode. It should be pointed out that in all mentioned samples the anodic peaks shifts in the direction of positive potential with increasing scan rate, which indicates the electrochemical doping/de-doping processes were chemically reversible for these samples.
The electroactivity behaviors of the synthesized samples were further studied by determination of the relationship between the peak current sizes versus scan rates and the results obtained are summarized in Fig. 9d. This linear relationship is typical of redox-active polymers attached to the electrodes and also exemplifies the stability of the synthesized samples toward doping/de-doping processes.
 | ||
| Fig. 10 UV-vis spectra of the pure PANI (a), PEG2000-b-(PANI)4 (b), and PEG6000-b-(PANI)4 (c) samples in DMSO solution. | ||
In similar, the UV-vis spectrum of the PEG2000-b-(PANI)4 sample (Fig. 10b) was characterized by two electronic transitions at about 315 and 613 nm, respectively. In contrast, the UV-vis spectrum of the PEG6000-b-(PANI)4 (Fig. 10c) was characterized by only one electronic transitions at about 601 nm, which related to the polaron formation and polaron to π* transition. In conclusion, the shifts to shorter wavelength (blue shift), especially in the case of PEG6000-b-(PANI)4 sample, are originated from decreasing of conjugated unit concentration by the PEG segments in comparison with pure PANI.
3.4. Characterization of electrospun nanofibers
As mentioned in Introduction section, an ideal scaffold should have some adequate physicochemical and biological characteristics (more especially mimicking of native ECM microenvironment), which have pivotal role in the performance of the scaffold during TE. Some of these properties were evaluated in the following sections. | ||
| Fig. 11 The SEM images of PEG2000-b-(PANI)4/PCL (a and b), and PEG6000-b-(PANI)4/PCL (c and d) electrospun nanofibers at different magnifications. | ||
The representative mechanical properties of the pure PCL, PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL electrospun nanofibers are shown in Fig. 12, and the results obtained are summarized in Table 1. According to Fig. 12, all fabricated nanofibers exhibited a linear elastic behavior before failure. As seen in Table 1, the PEG6000-b-(PANI)4/PCL has better mechanical properties in comparison with PEG2000-b-(PANI)4/PCL mainly due to higher amount of PEG. On the other hand, this phenomenon may be originated from higher molecular weight in the case of PEG6000.
 | ||
| Fig. 12 The stress–strain curves of pure PCL, PEG2000-b-(PANI)4/PCL, and PEG6000-b-(PANI)4/PCL electrospun nanofibers. | ||
| Sample | Young's modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|
| PCL | 246 ± 15.7 | 19.3 ± 1.12 | 137 ± 18.8 |
| PEG2000-b-(PANI)4/PCL | 170 ± 6.71 | 14.7 ± 0.82 | 60.2 ± 7.6 |
| PEG6000-b-(PANI)4/PCL | 178 ± 8.26 | 14.9 ± 0.75 | 78 ± 12.4 |
| Sample | Volume specific resistivity (ρ; Ω cm) | Electrical conductivity (σ; S cm−1) |
|---|---|---|
| a Electrospun nanofibers were fabricated as given in Experimental section. | ||
| PANI | 1.19 | 0.84 ± 0.07 |
| PEG2000-b-(PANI)4 | 1.74 | 0.57 ± 0.05 |
| PEG6000-b-(PANI)4 | 1.96 | 0.51 ± 0.05 |
| PEG2000-b-(PANI)4/PCLa | 342 | 0.003 ± 2 × 10−4 |
| PEG6000-b-(PANI)4/PCLa | 457 | 0.0022 ± 1.7 × 10−4 |
The surface hydrophilicites of PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL nanofibers were measured by water drop contact angle method at room temperature. Photographs of water drops onto the fabricated electrospun nanofibers are shown in Fig. 13 (top). The contact angles of PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL electrospun nanofibers with water drop were calculated to be 81 ± 3.1° and 76 ± 2.8°, respectively.
 | ||
| Fig. 13 The photographs of water drops on PEG2000-b-(PANI)4/PCL (a) and PEG6000-b-(PANI)4/PCL (b) electrospun nanofibers, and the SEM images of the PEG2000-b-(PANI)4/PCL (c) and PEG6000-b-(PANI)4/PCL (d) electrospun nanofibers after 15 days soaking in PBS. | ||
Biodegradation is a pivotal requirement of a scaffold for a successful TE application, in order to enable tissue integration and to avoid subsequent surgical removal of the scaffold. It is well established that the biodegradation rate of a polymeric scaffold depends on its some characteristics such as molecular weight, chemical structure and composition of (co-)polymer, hydrolytically unstable bonds, the level of hydrophilicity/hydrophobicity, glass transition temperatures (Tg), and crystalline/amorphous morphology.44–46
The in vitro degradability of the PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL nanofibers were investigated through evaluating the morphological change, and gravimetric measurements after soaking of nanofibers in phosphate-buffered saline (PBS; pH 7.4; Invitrogen, CA, USA) at 37 °C. The PBS was refreshed every five days. After reaching the desired time, the specimens were retrieved, washed several times with distilled water, dried in vacuum, and then weighed. The mass loss percentage was calculated from: (Wi − Wr)/Wi; where Wi and Wr are the initial and the residual dry weights of the nanofibers.
As can be seen from Fig. 14, both PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL electrospun nanofibers have a fast mass loss up to fourth week with a linear degradation trend, and after which the mass loss for both samples slows down. In addition, as seen in this figure, the PEG6000-b-(PANI)4/PCL sample showed a slightly higher in vitro degradability in comparison with PEG2000-b-(PANI)4/PCL sample. The mass loss for PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL nanofibers were calculated to be 36.4, and 41.7 wt%, respectively after six weeks.
 | ||
| Fig. 14 Degradation profiles of the PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL electrospun nanofibers in PBS. | ||
The in vitro degradability of the fabricated nanofibers was further investigated by means of SEM observation (Fig. 13; bottom). As seen in SEM images, both PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL samples were undergoing to swelling and degradation after 15 days incubating in PBS at 37 °C.
3.5. Biocompatibility
The biocompatibility of the scaffold and its degraded by-products are the fundamental requirements in all types of regenerative medicines, mainly due to its influences on cell attachment, proliferation, migration, differentiation, and formation of neo-tissue.47 Thus, the biocompatibility of scaffold must be approved before clinical therapy. The biocompatibility of the fabricated nanofibers were examined by assessing the adhesion, viability and proliferation of mouse fibroblast L929 cell line using SEM and MTT assay, respectively as discussed in the following sections. | ||
| Fig. 15 In vitro cytotoxicity effects of the PEG2000-b-(PANI)4/PCL (a) and PEG6000-b-(PANI)4/PCL (b) electrospun nanofibers on mouse fibroblast L929 cells ((c) negative control). | ||
 | ||
| Fig. 16 The mouse fibroblast L929 cells growth performances of the PEG2000-b-(PANI)4/PCL (a) and PEG6000-b-(PANI)4/PCL (b) electrospun nanofibers ((c) negative control). | ||
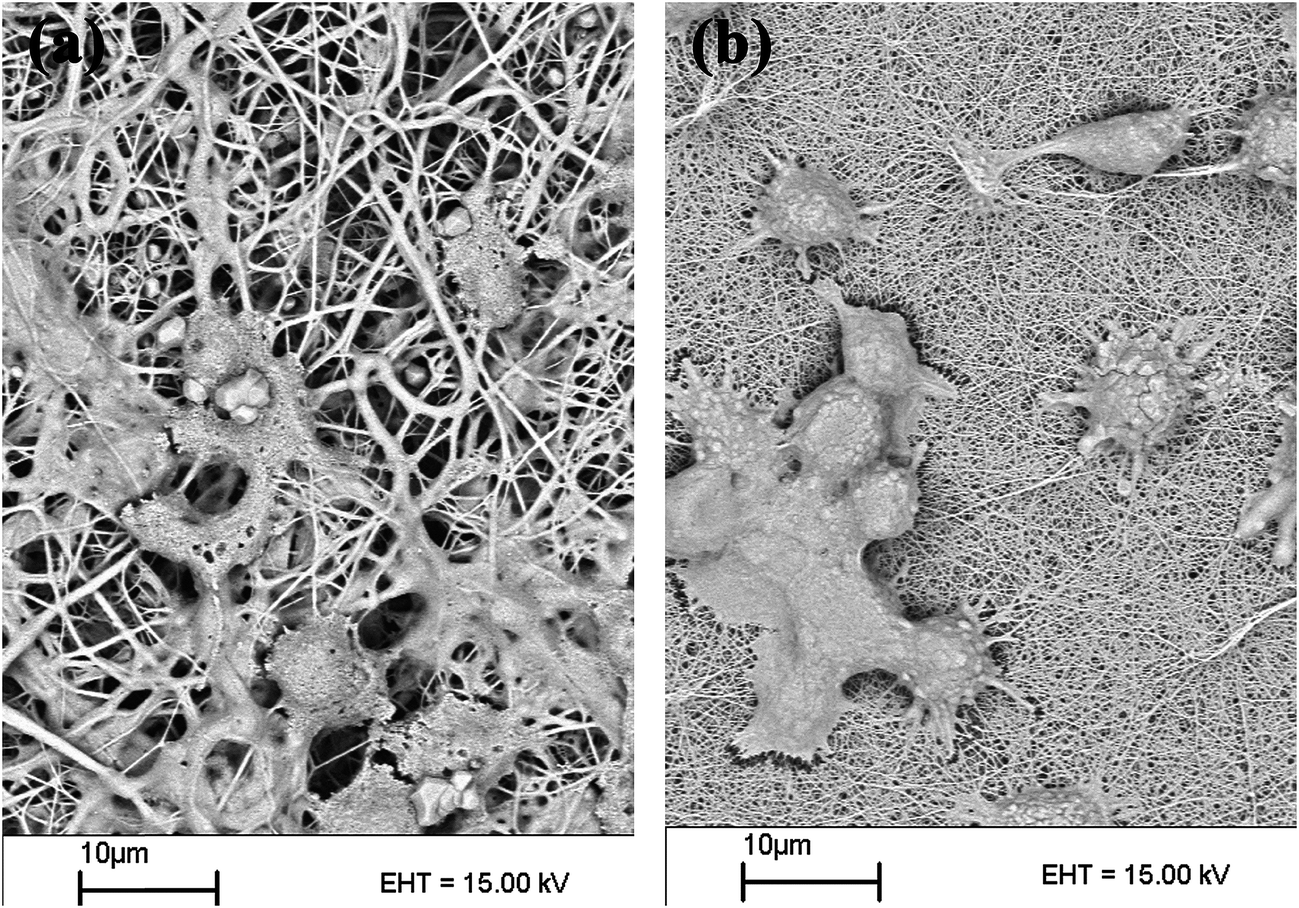 | ||
| Fig. 17 The SEM images of mouse fibroblast L929 cells on PEG2000-b-(PANI)4/PCL (a) and PEG6000-b-(PANI)4/PCL (b) electrospun nanofibers. | ||
4. Conclusion
The fabrication of two novel electrically conductive nanofibrous scaffolds based on poly(ethylene glycol)s-modified polyaniline [PEGs-b-(PANI)4] and poly(ε-caprolactone) (PCL) for tissue engineering (TE) applications were successfully demonstrated. The synthesis of PEG2000-b-(PANI)4 and PEG6000-b-(PANI)4 were verified by means of FTIR and 1H NMR spectroscopies, and their some physicochemical characteristics including morphologies, electroactivites, electrical conductivities, and thermal properties were also investigated. The fabricated PEG2000-b-(PANI)4/PCL and PEG6000-b-(PANI)4/PCL electrospun nanofibers showed uniform and 3D interconnected pore structure, with average diameters in the size range of 70 ± 30 nm. In addition, there are no beads formations were observed for both samples. Thus, these nanofibers could mimic the architecture and biological functions of the native ECM microenvironment. The biocompatibility of the fabricated scaffolds were primarily confirmed by evaluating the adhesion, viability and proliferation of mouse fibroblast L929 cells using SEM and MTT assay, respectively. As the conductivities, biocompatibilities, hydrophilicites, degradability's, and mechanical properties assessments results, we envision that the fabricated nanofibers could be potentially used as TE scaffolds that require electroactivity (e.g., skin, nerve, bone, and muscle reconstruction). In conclusion, further experiments are under progress in order to evaluate effects of electrical stimulation, composition, electrical conductivity and other physicochemical characteristics of the scaffolding biomaterials on the functional response of neural, fibroblast, and osteoblast cells.Acknowledgements
The authors are grateful to the Payame Noor University, University of Urmia, and Research Center for Pharmaceutical Nanotechnology, Tabriz University of Medical Sciences for partial financial support of this project. In addition, the authors sincerely thank Dr Ali Baradar-Khoshfetrat, Sahand University of Technology, Tabriz, Iran for his valuable technical and equipment's cooperation's.References
- F. M. Chen and X. Liu, Prog. Polym. Sci., 2016, 53, 86–168 CrossRef CAS PubMed.
- B. Maher, Nature, 2013, 499, 20–22 CrossRef CAS PubMed.
- M. V. Vellayappan, S. K. Jaganathan and E. Supriyanto, RSC Adv., 2015, 5, 73225–73240 RSC.
- J. Xie, M. R. MacEwan, S. M. Willerth, X. Li, D. W. Moran, S. E. Sakiyama-Elbert and Y. Xia, Adv. Funct. Mater., 2009, 19, 2312–2318 CrossRef CAS PubMed.
- E. A. Makris, A. H. Gomoll, K. N. Malizos, J. C. Hu and K. A. Athanasiou, Nat. Rev. Rheumatol., 2015, 11, 21–34 CrossRef CAS PubMed.
- B. M. Sicari, R. Londono and S. F. Badylak, J. Mater. Chem. B, 2015, 3, 7881–7895 RSC.
- T. Fee, C. Downs, A. Eberhardt, Y. Zhou and J. Berry, J. Biomed. Mater. Res., Part A, 2016, 104, 1680–1686 CrossRef CAS PubMed.
- E. Akaraonye, J. Filip, M. Safarikova, V. Salih, T. Keshavarz, J. C. Knowles and I. Roy, Polym. Int., 2016, 65, 780–791 CrossRef.
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- Y. Jia, W. Wang, X. Zhou, W. Nie, L. Chen and C. He, Polym. Chem., 2016, 7, 2553–2564 RSC.
- W. W. Thein-Han and H. H. K. Xu, Tissue Eng., Part A, 2013, 19, 1675–1685 CrossRef CAS PubMed.
- V. Vishwanath, K. Pramanik and A. Biswas, J. Biomater. Sci., Polym. Ed., 2016, 27, 657–674 CrossRef CAS PubMed.
- L. Li, J. Ge, L. Wang, B. Guo and P. X. Ma, J. Mater. Chem. B, 2014, 2, 6119–6130 RSC.
- J. H. Kim, D. K. Kim, O. J. Lee, H. W. Ju, J. M. Lee, B. M. Moon, H. J. Park, D. W. Kim, J. H. Lee and C. H. Park, Int. J. Biol. Macromol., 2016, 82, 160–167 CrossRef CAS PubMed.
- Y. Kang, G. Yin, Q. Yuan, Y. Yao, Z. Huang, X. Liao, B. Yang, L. Liao and H. Wang, Mater. Lett., 2008, 62, 2029–2032 CrossRef CAS.
- A. R. Jo, M. W. Hong, Y. S. Cho, K. M. Song and J. H. Lee, J. Appl. Polym. Sci., 2015, 132, 42566 CrossRef.
- C. V. C. Bouten, P. Y. W. Dankers, A. Driessen-Mol, S. Pedron, A. M. A. Brizard and F. P. T. Baaijens, Adv. Drug Delivery Rev., 2011, 63, 221–241 CrossRef CAS PubMed.
- T. A. Martin, S. R. Caliari, P. D. Williford, B. A. Harley and R. C. Bailey, Biomaterials, 2011, 32, 3949–3957 CrossRef CAS PubMed.
- Q. F. Guo, X. Li, Q. X. Ding, D. X. Li, Q. Zhao, P. Xie, X. H. Tang, F. Luo and Z. Qian, Int. J. Biol. Macromol., 2013, 58, 79–86 CrossRef CAS PubMed.
- S. Hinderer, S. L. Layland and K. Schenke-Layland, Adv. Drug Delivery Rev., 2016, 97, 260–269 CrossRef CAS PubMed.
- M. Jaymand, R. Sarvari, P. Abbaszadeh, B. Massoumi, M. Eskandani and Y. Beygi-Khosrowshahi, J. Biomed. Mater. Res., Part A, 2016, 104, 2673–2684 CrossRef CAS PubMed.
- B. Massoumi, R. Massoumi, N. Aali and M. Jaymand, J. Polym. Res., 2016, 23, 136 CrossRef.
- B. Song, Y. Gu, J. Pu, B. Reid, Z. Zhao and M. Zhao, Nat. Protoc., 2007, 2, 1479–1489 CrossRef CAS PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Tissue Eng., Part B, 2013, 19, 48–57 CrossRef CAS PubMed.
- J. C. Ojingwa and R. R. Isseroff, J. Invest. Dermatol., 2003, 121, 1–12 CAS.
- P. Petrov, P. Mokreva, I. Kostov, V. Uzunova and R. Tzoneva, Carbohydr. Polym., 2016, 140, 349–355 CrossRef CAS PubMed.
- K. D. McKeon-Fischer and J. W. Freeman, J. Tissue Eng. Regener. Med., 2011, 5, 560–568 CrossRef CAS PubMed.
- O. Akhavan, J. Mater. Chem. B, 2016, 4, 3169–3190 RSC.
- R. Sarvari, B. Massoumi, M. Jaymand, Y. Beygi-Khosrowshahi and M. Abdollahi, RSC Adv., 2016, 6, 19437–19451 RSC.
- M. Gizdavic-Nikolaidis, S. Ray, J. R. Bennett, A. J. Easteal and R. P. Cooney, Macromol. Biosci., 2010, 10, 1424–1431 CrossRef CAS PubMed.
- K. M. Persson, R. Karlsson, K. Svennersten, S. Löffler, E. W. H. Jager, A. Richter-Dahlfors, P. Konradsson and M. Berggren, Adv. Mater., 2011, 23, 4403–4408 CrossRef CAS PubMed.
- M. Jaymand, M. Hatamzadeh and Y. Omidi, Prog. Polym. Sci., 2015, 47, 26–69 CrossRef CAS.
- M. Gizdavic-Nikolaidis, J. Travas-Sejdic, G. A. Bowmaker, R. P. Cooney, C. Thompson and P. A. Kilmartin, Curr. Appl. Phys., 2004, 4, 347–350 CrossRef.
- R. Ravichandran, S. Sundarrajan, J. R. Venugopal, S. Mukherjee and S. Ramakrishna, J. R. Soc., Interface, 2010, 7, S559–S579 CrossRef CAS PubMed.
- B. Massoumi, N. Aali and M. Jaymand, RSC Adv., 2015, 5, 107680–107693 RSC.
- B. Massoumi, N. Sorkhi-Shams, M. Jaymand and R. Mohammadi, RSC Adv., 2015, 5, 21197–21205 RSC.
- B. Massoumi, V. Badr-Valizad and M. Jaymand, RSC Adv., 2015, 5, 40840–40848 RSC.
- B. Massoumi, N. Poorgholy and M. Jaymand, Int. J. Polym. Mater. Polym. Biomater., 2017, 1, 38–47 CrossRef.
- T. H. Qazi, R. Rai, D. Dippold, J. E. Roether, D. W. Schubert, E. Rosellini, N. Barbani and A. R. Boccaccini, Acta Biomater., 2014, 10, 2434–2445 CrossRef CAS PubMed.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS.
- B. Massoumi, M. Ramezani, M. Jaymand and M. Ahmadinejad, J. Polym. Res., 2015, 22, 214 CrossRef.
- E. A. Rainbolt, K. E. Washington, M. C. Biewer and M. C. Stefan, Polym. Chem., 2015, 6, 2369–2381 RSC.
- B. Massoumi, R. Mohammad-Rezaei and M. Jaymand, Polym. Adv. Technol., 2016, 27, 1056–1063 CrossRef.
- J. Kopeček and K. Ulbrich, Prog. Polym. Sci., 1983, 9, 1–58 CrossRef.
- C. E. Leblon, R. Pai, C. R. Fodor, A. S. Golding, J. P. Coulter and S. S. Jedlicka, J. Appl. Polym. Sci., 2013, 128, 2701–2712 CrossRef CAS.
- S. Sant, D. Iyer, A. K. Gaharwar, A. Patel and A. Khademhosseini, Acta Biomater., 2013, 9, 5963–5973 CrossRef CAS PubMed.
- Y. Yadong, L. Xiaoying and D. Fei, J. Biomed. Nanotechnol., 2015, 11, 816–827 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |