
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver comes into play: Henry reaction and domino cycloisomerisation sequence catalysed by [Ag(I)(Pc-L)] complexes†
Giorgio Tseberlidisa,
Monica Dell'Acquab,
Daniele Valcarenghia,
Emma Galloa,
Elisabetta Rossib,
Giorgio Abbiati *b and
Alessandro Caselli*a
*b and
Alessandro Caselli*a
aDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19 – 20133, Milano, Italy. E-mail: alessandro.caselli@unimi.it
bDipartimento di Scienze Farmaceutiche, Sezione di Chimica Generale e Organica “A. Marchesini”, Università degli Studi di Milano, Via Venezian, 21–20133, Milano, Italy. E-mail: giorgio.abbiati@unimi.it
First published on 6th October 2016
Abstract
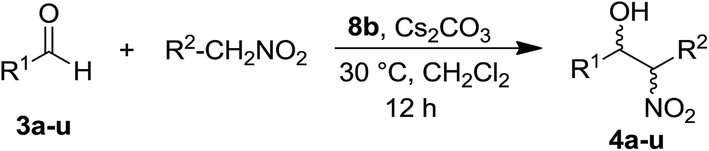
We report herein the synthesis of new pyridine-containing macrocyclic ligands (Pc-L) bearing a non-innocent pendant arm, by exploiting both chiral and functional properties of natural amino acids. The obtained macrocyclic ligands were employed to synthesize well-defined cationic silver(I) complexes that were shown to be competent catalysts for the Henry (nitroaldol) reaction. Good to excellent yields and full selectivity in the β-nitroalcohol product were obtained starting from electron-poor aromatic aldehydes or other activated aldehydes such as furfural under mild reaction conditions. The straightforward synthesis of the macrocyclic ligands starting from cheap commercially available starting materials allowed the introduction of a suitable basic functionality into the ligand pendant arm, thus providing a bifunctional catalyst. Based on our previous experience in the [Ag(I)(Pc-L)] catalysed domino addition/cycloisomerisation reaction of o-alkynylbenzaldehydes and nucleophiles, the synthesis of isochromenes coupling the Henry reaction and the cycloisomerisation in a single step was subsequently explored. Although with low selectivity, [Ag(I)(Pc-L)] cationic complexes were able to promote such a cascade reaction and a possible mechanism based on experimental evidence has been proposed.
Introduction
Carbon–carbon bond forming reactions, due to their unique capability of generating molecular complexity, represent a powerful tool in the reserves of a synthetic chemist.1,2 The last decade has witnessed considerable advances in the catalytic generation and enantioselective addition of carbon nucleophiles (i.e., enolates3 and nitronates4) to different types of electrophiles. In this context, nitroalkanes are important reagents not only due to their propensity to undergo easy α-dehydrogenation but also for their facile interconversion to other organic functional groups.5,6 Even weak bases are capable of abstracting a proton in the α-position of a nitro group (pKa ∼ 10) and the nucleophilic attack of the generated nitronate anion on a carbonyl compound to give a β-nitro alcohol is referred to as the Henry (or nitroaldol) reaction.7 Although more than one century old, this reaction is still to be considered one of the most important examples of an atom-economical transformation.8–10 No need for a stoichiometric amount of a base, metal-catalysts,11–15 enzymes16,17 or organocatalysts18–20 can efficiently promote the Henry reaction. Among the metal complexes commonly employed as catalysts, copper(I)21–30 and copper(II)11,31–44 complexes play a prominent role, whereas, to the best of our knowledge the activity of silver salts and complexes have rarely been tested. In several cases reported in the literature, the replacement of Cu with Zn gave comparable results but in some of them a reversed enantioselectivity was observed.27,32,33,42,45 Cu-catalysed reactions are believed to proceed by a monometallic form of active species whereas there is strong evidence of the involvement of a multimetallic species as actual catalyst in Zn(II) promoted Henry reactions.26 Surprisingly, such a correlation in reactivity with silver has never been made and the few examples appeared in the literature report that silver salts either failed in promoting the reaction,40,46 or gave very poor yields.36,47In the past few years, our attention has turned to the introduction of a pyridine moiety into the skeleton of tetraaza-macrocycles, with the aim to obtain ligands with increased conformational rigidity and different basicity.48 The copper(I) complexes of these 12-membered pyridine-containing ligands (Pc-L, Fig. 1) have been successfully employed as catalysts in the enantioselective cyclopropanation of alkenes.49–52 The same Cu(I)-catalysts have shown good activities in the Henry reaction.22 Among coinage metals, reports on the catalytic activity of silver complexes are relatively sparse when compared to the more extensively studied copper and gold.53–59 We have recently reported the full characterization of [silver(I)(pyridine-containing ligand)] complexes and their organometallic reactivity,60 and we have demonstrated their catalytic activity in the regiospecific domino synthesis of 1-alkoxy-isochromenes under mild conditions61 and in the microwave enhanced A3-coupling multicomponent reaction.62 Compared with simple silver salts, the great advantages of [Ag(I)(Pc-L)] complexes are their solubility, their enhanced stability and the easiness of handling. Prompted by the interesting results above mentioned, we were intrigued to check if our well-defined silver(I) complexes were suitable catalysts also for the Henry reaction. As a result of this study, we were pleased to find that [Ag(I)(Pc-L)] complexes can actually activate the aldehyde toward the nitronate nucleophilic attack, in the first example of a silver mediated Henry reaction. Since the Henry reaction, especially in the asymmetric version, has provided a good platform for testing the dual activation of metal/base catalysts,63 we have modified our ligands in order to attach in the proper position a suitable base to facilitate the reaction. Moreover, in connection with our ongoing interest in the study of domino nucleophilic addition/cyclization reaction involving alkynes characterized by the presence of a proximate nucleophile,64–69 we envisaged to test the reaction on proper bifunctional substrates. Based on our experience, we chose the o-alkynylarylaldehydes with the ambition to join the Ag catalysed Henry reaction (by activation of the aldehyde moiety) to the Ag catalysed cycloisomerisation (by activation of the triple bond) with the aim to obtain in a cascade fashion new interesting O-heterocycles such as isochromenes.
 | ||
| Fig. 1 Pyridine-containing ligand (Pc-L*) and numbering scheme adopted. | ||
Results and discussion
Silver catalysed Henry reaction
At first, we decided to test the ability of silver to promote the Henry reaction by using simple silver(I) salts such as Ag(OTf) or Ag(BF4) and the Pc-L silver(I) complex 160 (already used in the A3-coupling study), and to compare their reactivity with those of the corresponding Pc-L copper(I) complex 250 (Fig. 2). As model reaction, we chose the condensation between 4-nitrobenzaldehyde 3a and nitromethane (Table 1). To promote the generation of the nitronate anion, a catalytic amount of triethylamine (TEA) – equal to the catalyst loading – was added to the reaction mixture.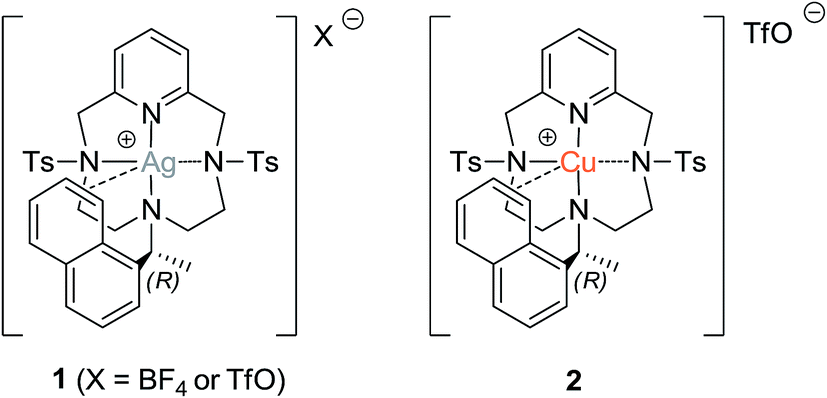 | ||
| Fig. 2 Silver (1) and copper (2) PcL-complexes tested in the model Henry reaction. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | X | Solv. | Base | Yieldb (%) |
a Reactions were performed with [Ag(I)] (3.2 × 10−2 mmol) in the solvent (5 mL) at a cat./base/aldehyde/nitromethane ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 : 10:50 at rt for 20 h; lower catalyst loadings (1 mol%), resulted in very slow reactions.b Isolated yields based on initial 4-nitrobenzaldehyde; unreacted aldehyde accounted for the rest of the reaction mass balance.c T = 30 °C, t = 6 h.d Reaction conditions in the absence of metal catalyst: TEA/aldehyde/nitromethane ratio of 1:10:500 at rt for 20 h. Unreacted 4-nitrobenzaldehyde did not account for the rest of the reaction mass balance and some unidentified by-products derived from competitive side reactions were observed.e In the presence of molecular sieves (4 Å). :1 : 10:50 at rt for 20 h; lower catalyst loadings (1 mol%), resulted in very slow reactions.b Isolated yields based on initial 4-nitrobenzaldehyde; unreacted aldehyde accounted for the rest of the reaction mass balance.c T = 30 °C, t = 6 h.d Reaction conditions in the absence of metal catalyst: TEA/aldehyde/nitromethane ratio of 1:10:500 at rt for 20 h. Unreacted 4-nitrobenzaldehyde did not account for the rest of the reaction mass balance and some unidentified by-products derived from competitive side reactions were observed.e In the presence of molecular sieves (4 Å). |
|||||
| 1 | Ag(OTf) | CH2Cl2 | TEA | 21 | |
| 2c | Ag(BF4) | CH2Cl2 | Cs2CO3 | 25 | |
| 3d | — | CH2Cl2 | TEA | 35 | |
| 4 | 1-(13R) | OTf | CH2Cl2 | TEA | 84 |
| 5 | 1-(13R) | BF4 | CH2Cl2 | TEA | 75 |
| 6e | 1-(13R) | BF4 | CH2Cl2 | TEA | 85 |
| 7 | 1-(13R) | OTf | CH3NO2 | TEA | 84 |
| 8 | 1-(13R) | OTf | Tol | TEA | 60 |
| 9 | 2-(13R) | OTf | CH2Cl2 | TEA | 85 |

Simple silver(I) salts gave results similar to those observed just in the presence of the base, the only difference being that in the latter case a number of by-products were observed (Table 1, compare entries 1, 2 and 3). On the other hand, complex 1, which is fully soluble in chlorinated solvents, gave results comparable to those obtained with the related copper complex 2 (Table 1, entries 4 and 9). Under these conditions, the reaction gave identical results either employing the preformed complex 1 or the in situ formed 1:1 ligand/Ag(I) complex. We briefly investigated also the role of the counteranion, by changing from OTf− to BF4−, obtaining a slightly lower yield with the latter (Table 1, entry 5). However, when the same reaction was repeated in the presence of molecular sieves, a comparable conversion was again obtained (Table 1, entry 6). The use of a large excess of nitromethane (Table 1, entry 7) did not affect the reaction yield. We previously observed that the copper Pc-L complexes failed to catalyse the Henry reaction when it was performed in aromatic hydrocarbons and that Pc-L ligands alone, in the absence of any metal, are not suitable catalysts.22 On contrary, the silver Pc-L complex 1 demonstrated to be effective also in toluene, giving the desired nitroalcohol 4a in 60% yield (Table 1, entry 8). Finally, despite the presence of a defined stereocentre on the ligand, under all conditions tested we ever obtained the nitroalcohol 4a as a racemic mixture.
Optimization of the silver(I) catalysts for the Henry reaction
Having in hand a quite active catalytic system, we were intrigued to further optimize the ligand features and then to explore the scope and limitations of the title reaction. The Pc-L's are easily constructed in good to excellent yields taking advantage of a 2 + 2 Richman–Atkins-type coupling,70 which has been modified in order to improve chemical yields, as already reported.50 In order to extend our method to the synthesis of Pc-L's bearing a non-innocent pendant arm on N6 (i.e., a pendant with coordinating, basic or acidic functionalities), we envisioned that natural amino acids (i.e., β-alanine and lysine) could be perfect nucleophilic reaction partners for the ring opening reaction with 2 moles of N-tosyl aziridine to give the corresponding 4-substituted 1,4,7-triazaheptanes 5a,b, as depicted in Scheme 1.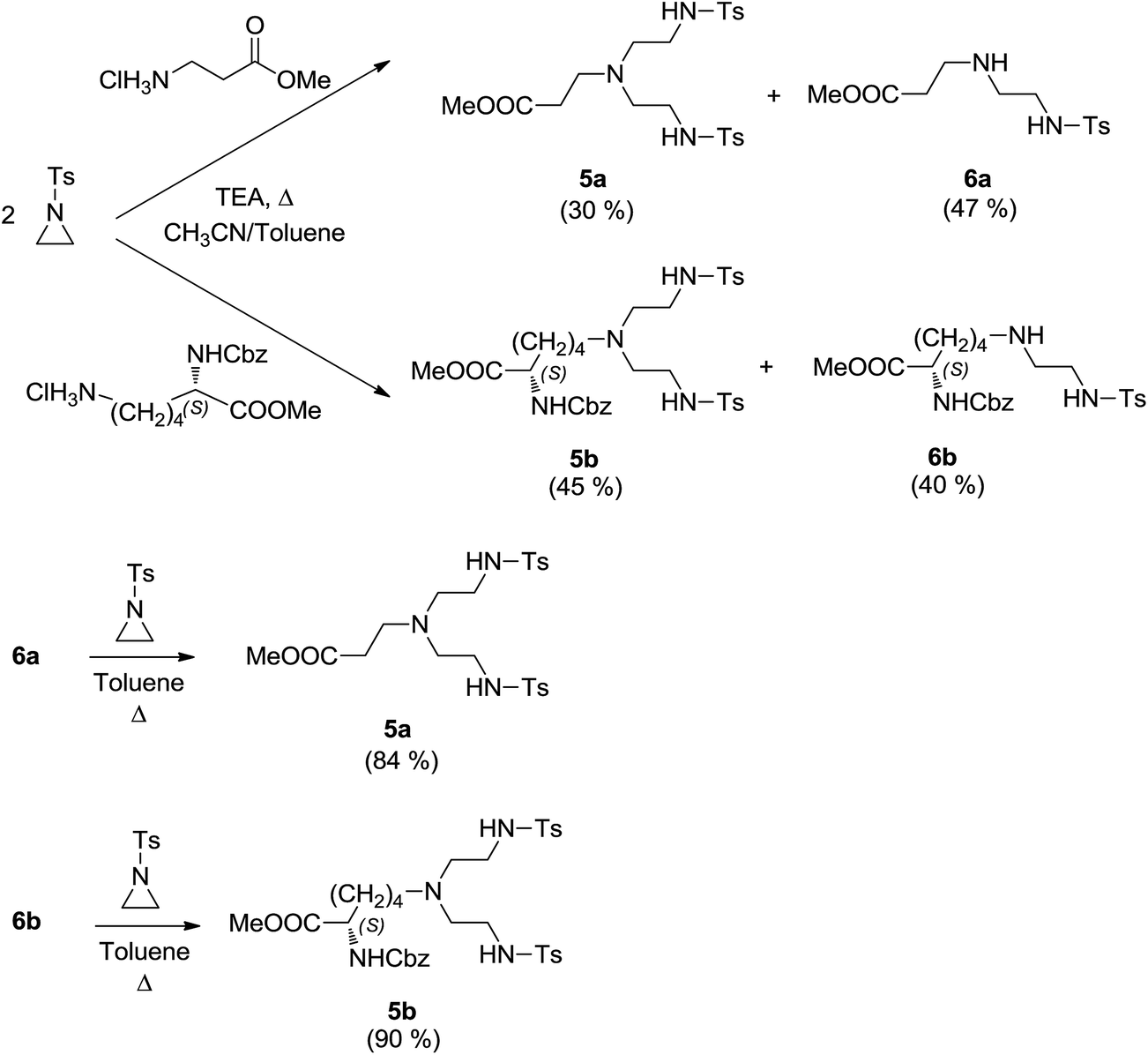 | ||
| Scheme 1 Synthesis of amino acid derived 4 substituted 1,4,7-triazaheptanes 5a and 5b. | ||
In particular, β-alanine methyl ester hydrochloride and (S)-N(α)-Cbz-lysine methyl ester hydrochloride were quantitatively obtained by esterification of the corresponding amino acid with methanol in the presence of trimethylchlorosilane at room temperature.71 Subsequent reaction with tosyl protected aziridine in a refluxing mixture of CH3CN/toluene in the presence of TEA resulted in the formation of desired bis(sulfonamides) 5a–b in quite fair yields, along with the corresponding mono adducts 6a–b. However, after chromatographic purification, isolated mono(sulfonamides) 6a–b were easily converted to 5a–b in very good yields (Scheme 1).
The following Richman–Atkins cyclisation with 2,6-pyridinedimethanol 2,6-dimesylate61 yielded to the 12-membered macrocycles 7a–b in excellent yields (Scheme 2). Catalytic hydrogenation of 7b allowed the clean and quantitative removal of the Cbz amino-protecting group to afford 7c, which was converted to macrocycle 7d, containing a basic tertiary amine pendant arm, by reductive amination in presence of acetaldehyde and sodium cyanoborohydride (Scheme 2). To check the stability of the original stereocentre of the starting lysine under the reactions conditions, the optical rotation of the intermediates 5–7b was measured after each reaction step. The values recorded are consistent with a complete retention of configuration, as confirmed via 19F NMR analysis of the Mosher's amide formation with ligand 7c (see ESI†). To further confirm this data, ligand 7c was reacted in deuterated chloroform with both enantiomers of the α-methoxyphenylacetic acid in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP). In both cases, a single diastereoisomer was obtained (see Experimental section and ESI†).
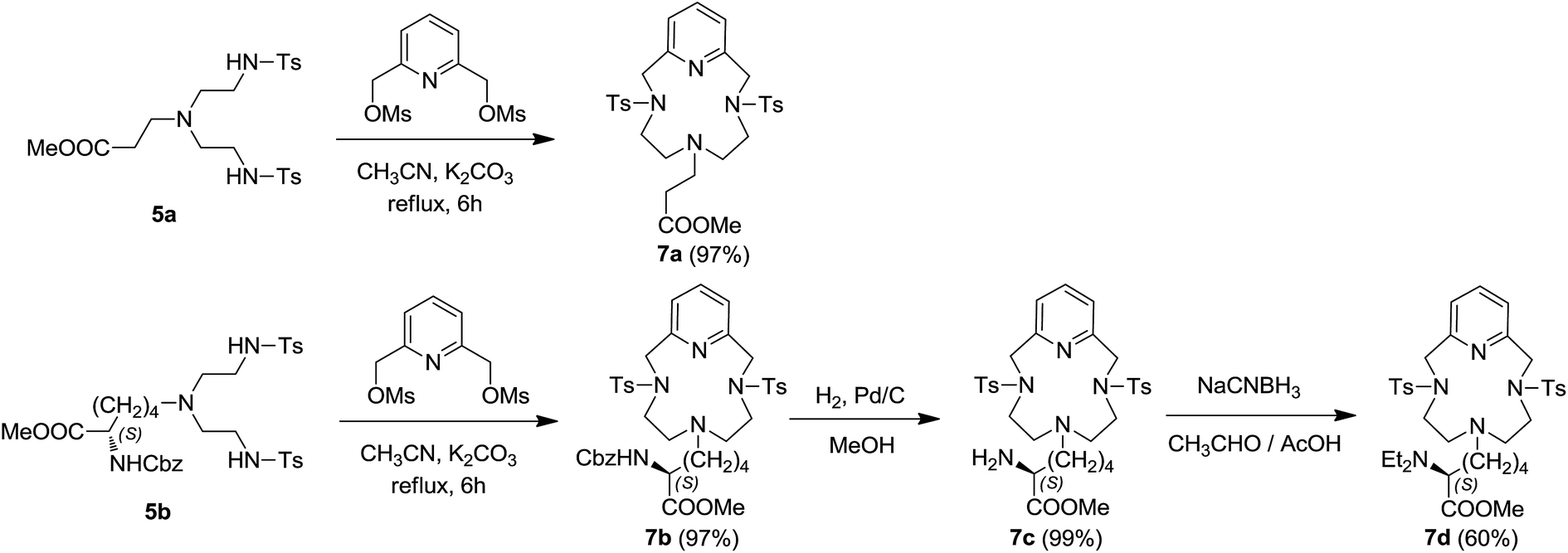 | ||
| Scheme 2 Synthesis of Pc-L's 7a–d. | ||
Metal complex formation with ligands 7a–d has been performed both with Ag(OTf) and Ag(BF4), by treating at room temperature a solution of the ligand in dichloroethane (DCE) under nitrogen atmosphere (Scheme 3). After filtering off eventual uncomplexed metal salt, the colourless solution was concentrated, and the addition of n-hexane favoured the formation of a white precipitate of [Ag(Pc-L)] complex in good to excellent yields. All complexes have been isolated and fully characterised. They showed a remarkable stability both in solid and in solution phase and can be handled in air without decomposition even for a prolonged period.
 | ||
| Scheme 3 Synthesis of silver(I) complexes 8a–d. | ||
The synthesis of these ligands is relatively simple and high-yielding. Moreover, the major advantage of this synthetic approach is that it allows an easy modification of a key moiety of the macrocycle, namely the N6 pendant, by exploiting both chiral and functional properties of the natural amino acids. The new Ag(I) complexes 8a–d, where tested as catalysts in the model Henry reaction between 4-nitrobenzaldehyde 3a and nitromethane. According to the best results obtained in the preliminary screening, the reactions where performed in CH2Cl2 with a ratio cat/base/aldehyde/CH3NO2 of 1:1:10:50. As during the optimization studies we noticed that adventitious traces of water could in some cases affect the reproducibility of the results, all new reactions have been carried out in the presence of activated molecular sieves. The results are reported in Table 2.
| Entry | Cat. | X | Base | Solv. | t (h) | T (°C) | Yieldb 4a (%) |
|---|---|---|---|---|---|---|---|
| a Reactions were performed with [Ag(I)] (3.2 × 10−2 mmol) in the solvent (5 mL) at a cat./base/aldehyde/nitromethane ratio of 1:1:10:50 in the presence of molecular sieves (4 Å).b Yields based on initial 4-nitrobenzaldehyde calculated via 1H NMR using 2,4-dinitrotoluene (DNT) as internal standard; unreacted aldehyde accounted for the rest of the reaction mass balance.c The metal complex 8c is not soluble in the reaction medium: no reaction after 20 h as judged by TLC analysis.d Dimethyl acetal derived from the nucleophilic attack of MeO− on the 4-nitrobenzaldehyde was recovered in 25% yield (see ESI). |
|||||||
| 1 | 8a | OTf | TEA | CH2Cl2 | 20 | rt | 60 |
| 2 | 8a | OTf | TEA | Tol | 40 | rt | 15 |
| 3 | 8a | OTf | DiPEA | CH2Cl2 | 20 | rt | 53 |
| 4 | 8a | OTf | DMAP | CH2Cl2 | 20 | rt | 53 |
| 5 | 8a | OTf | Morpholine | CH2Cl2 | 20 | rt | 50 |
| 6 | 8a | OTf | K2CO3 | CH2Cl2 | 20 | rt | 45 |
| 7 | 8a | OTf | Cs2CO3 | CH2Cl2 | 20 | rt | 70 |
| 8 | 8a | BF4 | Cs2CO3 | CH2Cl2 | 20 | rt | 70 |
| 9 | 8b | OTf | TEA | CH2Cl2 | 20 | rt | 67 |
| 10 | 8b | BF4 | TEA | CH2Cl2 | 20 | rt | 65 |
| 11 | 8b | BF4 | DiPEA | CH2Cl2 | 20 | rt | 75 |
| 12 | 8b | BF4 | Cs2CO3 | CH2Cl2 | 20 | rt | 90 |
| 13 | 8b | BF4 | Cs2CO3 | CH2Cl2 | 20 | 30 | 95 |
| 14 | 8b | BF4 | Cs2CO3 | CH2Cl2 | 12 | 30 | 92 |
| 15 | 8b | BF4 | Cs2CO3 | CH2Cl2 | 6 | 30 | 75 |
| 16 | 8b | BF4 | Cs2CO3 | CH2Cl2 | 3 | 30 | 45 |
| 17 | 8b | OTf | — | CH2Cl2 | 20 | rt | 25 |
| 18 | — | — | Cs2CO3 | CH2Cl2 | 20 | 30 | 55 |
| 19c | 8c | BF4 | — | CH2Cl2 | 20 | rt | — |
| 20c | 8c | BF4 | — | MeNO2 | 20 | rt | — |
| 21 | 8c | BF4 | — | MeOH | 20 | rt | —d |
| 22 | 8d | BF4 | Cs2CO3 | CH2Cl2 | 12 | 30 | 90 |
| 23 | 8d | BF4 | — | CH2Cl2 | 12 | 30 | 64 |
Except for complex 8c, which is the only one not soluble in dichloromethane, all other silver complexes gave the nitroalcohol 4a from moderate to very good yields in the model reaction. Beside the desired nitroalcohol 4a the starting aldehyde 3a was the only product recovered at the end of the reaction, thus yields reported are coincident with conversions and a selectivity >99% was observed.
Silver complex 8a (Table 2, entries 1–8), characterised by the presence of a slightly coordinating N6 pendant arm, gave on the whole results comparable to those observed by using complex 1. Under the previously optimised reaction conditions, the reaction yield is slightly lower (cfr. Table 1, entry 4 and Table 2, entry 1). The use of a less polar solvent resulted in a drop of the reaction yield (Table 2, entry 2). Several bases were tested (Table 2, entries 3–7), and best results were obtained with cesium carbonate (Table 2, entry 7). The counter anion do not seem to influence the reaction yields (Table 2, entry 8), in agreement with the 1H NMR spectra of the 8a-BF4 and 8a-OTf that do not show any apparent interaction between the counterion and the metal complex (see ESI†).
Complex 8b, characterised by the presence of a Cbz-protected amino group on the N6 pendant arm, displayed a closely related behaviour regarding counter anion (Table 2, entries 9 and 10) and base (Table 2, entries 10–12) effects, but overall, the yields were higher. With catalyst 8b, the best results were obtained in the presence of 1 eq. of Cs2CO3 in dichloromethane (Table 2, entries 12–14): after 20 h at rt, 4a was formed in 90% yield (Table 2, entry 12); a rise in temperature to 30 °C resulted in an increase of yield to 95% (Table 2, entry 13), whereas a reduction of reaction time gave gradually worse results (Table 2, entries 14–16). As expected, in the absence of a base, poor results were obtained (Table 2, entry 17). It should be pointed out that Cs2CO3 alone is able to promote the Henry reaction, but under the same reaction conditions only a 55% of nitroalcohol 4a was formed (Table 2, entry 18).
In order to avoid the addition of the base as co-catalyst, we tested complexes 8c and 8d, both containing a basic amine functionality on the N6 active-pendant arm.
As above pointed out, complex 8c is insoluble in dichloromethane, as well as in nitromethane and failed to give any reaction in such heterogeneous system (Table 2, entries 19 and 20). When methanol was used as solvent, we observed the formation of the dimethyl acetal derived from the nucleophilic attack of MeO− on the 4-nitrobenzaldehyde in 25% yield (Table 2, entry 21).
Under standard conditions, complex 8d, which is fully soluble in dichloromethane, gave the Henry product 4a in excellent yields (Table 2, entry 22). Moreover, we were pleased to observe that the presence of a tertiary amino group on the N6 active-pendant made this complex able to promote the formation of 4a in 64% yields also without any basic co-catalyst in just 12 h at 30 °C (Table 2, entry 23). Unfortunately, again, despite the presence of a stereocentre in S configuration close to the amine, the enantioinduction in the product is very low (5% ee determined by chiral HPLC).
As already pointed out, the presence of NH2 and/or COOH functional groups in the ligands could allow for the synthesis of di- or oligo-peptides embedding the metal complex. Hopefully, the chirality imposed by a more extended peptide structure to the whole catalytic system will provide a more stereoselective transition state for the two incoming reaction partners. Current efforts in our laboratory are now devoted in this direction with the aim to improve the enantioselective outcome in this new silver catalysed Henry reaction.
We next explored scope and limitation of the approach (Table 3), employing complex 8b-BF4 under the best reaction conditions (Table 2, entries 13 and 14). In particular, our interest was to verify the ability of our catalytic system to promote the reaction of aldehydes of different nature (aryl, heteroaryl and cycloalkyl) also in the presence of EW or ED groups on the aromatic ring. We decide to perform all reactions in CH2Cl2 at 30 °C and stop them after 12 h with the aim to compare the results obtained by changing the electrophilic properties of the aldehyde at fixed reaction time and temperature. It is well known that the Henry reaction is an equilibrium reaction and temperature plays an important role. The 12 h reaction time was decided based on a brief kinetic study performed on the model reaction (Table 2, entries 13–16), which displayed that a plateau in terms of % of conversion of the starting aldehyde is reached just after 12 h (Fig. 3).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | syn/antib | Yieldc (%) |
| a Reactions were performed with [Ag(I)] (3.2 × 10−2 mmol) in CH2Cl2 (5 mL) at a cat./Cs2CO3/aldehyde/nitromethane ratio of 1:1:10:50 in the presence of molecular sieves (4 Å) at 30 °C for 12 h.b syn/anti ratio determined by 1H NMR.c Yields based on initial aldehyde calculated via 1H NMR using 2,4-dinitrotoluene (DNT) as internal standard (isolated yields); unreacted aldehyde accounted for the rest of the reaction mass balance.d Reaction performed with complex 8a-BF4 as catalyst.e Reaction performed with complex 8d-BF4 as catalyst and in the absence of Cs2CO3. |
|||||
| 1 | 4-NO2C6H4 | H | 4a | — | 92 (90) |
| 2 | 4-CNC6H4 | H | 4b | — | 83 (80) |
| 3 | 4-CF3C6H4 | H | 4c | — | 93 (92) |
| 4 | 4-BrC6H4 | H | 4d | — | 85 (82) |
| 5 | 4-ClC6H4 | H | 4e | — | 83 (80) |
| 6 | 4-FC6H4 | H | 4f | — | 70 (64) |
| 7 | C6H5 | H | 4g | — | 60 (55) |
| 8d | 4-MeC6H4 | H | 4h | — | 15 |
| 9 | 4-ButC6H4 | H | — | — | n.d. |
| 10 | 4-MeOC6H4 | H | — | — | n.d. |
| 11 | 4-Et2NC6H4 | H | — | — | n.d. |
| 12 | C6F5 | H | 4i | — | 72 (68) |
| 13 | 3,5-(CF3)2C6H4 | H | 4j | — | 70 (66) |
| 14 | 2-NO2C6H4 | H | 4k | — | 97 (95) |
| 15 | 2-BrC6H4 | H | 4l | — | 72 (65) |
| 16 | 2,6-Cl2C6H4 | H | 4m | — | 70 (68) |
| 17 | 2-MeOC6H4 | H | 4n | — | 10 |
| 18 | 3-MeOC6H4 | H | — | — | n.d. |
| 19 | Cy | H | — | — | n.d. |
| 20 |  |
H | — | — | n.d. |
| 21 |  |
H | — | — | n.d. |
| 22 |  |
H | 4o | — | 65 (62) |
| 23 |  |
H | 4p | — | 10 |
| 24 | 4-NO2C6H4 | CH3 | 4q | 60:40 |
95 (90) |
| 25e | 4-NO2C6H4 | CH3 | 4q | 60:40 |
90 |
| 26 | 2-NO2C6H4 | CH3 | 4r | 45:55 |
96 (93) |
| 27 | 4-CF3C6H4 | CH3 | 4s | 51:49 |
93 (89) |
| 28 | 4-ClC6H4 | CH3 | 4t | 61:39 |
80 (75) |
| 29 | C6H5 | CH3 | 4u | 55:45 |
75 (60) |
 | ||
| Fig. 3 Conversion of 3a vs. time in the Henry reaction catalysed by 8b-BF4/Cs2CO3 in CH2Cl2 at 30 °C. | ||
Good results in terms of yields were obtained when one or more EWGs were present on the aryl ring (Table 3, entries 1–6 and 12–16). Although not linearly correlated with σpara Hammet constants, yields between 83% and 93% were obtained with aromatic aldehydes having high positive values of para substituent constant (Table 3, entries 1–5). Slightly lower yields have been observed for para- and per-fluorobenzaldehydes (Table 3, entries 6 and 12), and neutral benzaldehyde (Table 3, entry 7). On the other hand, electron-rich aromatic aldehydes failed to give the Henry products (Table 3, entries 8–11). These latter results can be easily rationalised based on the less pronounced electrophilic character of the aldehyde. The presence of EW groups is well tolerated also in meta and ortho positions (Table 2, entries 13–16), suggesting that steric hindrance does not limit the transformation.
As observed for electron-rich benzaldehydes, no reaction was observed with aliphatic aldehydes (Table 3, entry 19) and with cinnamaldehyde (Table 3, entry 20). Among furan, tiophene and pyrrole carbaldehydes, a good reactivity was observed only in the case of furfural (Table 3, entry 22), the less “aromatic” among the five-membered heterocycles, in which, probably, the strong inductive EW effect of the oxygen atom plays a key role in the activation of the adjacent aldehyde functionality. On contrary, the reactions of thiophene-2-carbaldehyde and N-methylpyrrole-2-carbaldehyde failed (Table 3, entries 21 and 23). In particular, the very poor yield obtained in the reaction of thiophene-2-carbaldehyde (Table 3, entry 23) can be also related to a plausible strong coordination of the sulphur atom to the metal centre that inhibits any catalytic activity, whilst in the case of N-methylpyrrole-2-carbaldehyde (Table 3, entry 21) only a mixture of unidentified by-products, probably derived from polymerization, were observed.
Next, we briefly explored the reactivity of some aromatic aldehydes with nitroethane, with a particular regard to the diastereoselective outcome of the reaction. In all cases, almost identical results were obtained switching the nitro partner from nitromethane to nitroethane, and all the reactions with electron poor aromatic aldehydes gave the corresponding Henry product in very good yields, although with very modest syn/anti ratio (Table 3, entries 24–29). Again, we were pleased to verify that complex 8d-BF4 was able to catalyse the reaction between 4-nitrobenzaldehyde and nitroethane without the need of any additional base, yielding 4q in a very satisfying 90% yield (Table 3, entry 25).
Taking into account that the indole nucleus is present in a large number of compounds of biological and/or pharmaceutical interest, we tested our approach also on isatine (Scheme 4).
 | ||
| Scheme 4 8b-BF4 catalysed reaction of nitromethane and nitroethane with isatine. | ||
The reaction with nitromethane gave the desired 3-hydroxy-3-(nitromethyl)-1,3-dihydro-2H-indol-2-one 9a in 80% isolated yield72 whereas the reaction with nitroethane gave a 6:4 diastereoisomeric mixture of 9b with an overall yield of 97% (yield based on initial isatine calculated via 1H NMR using 2,4-dinitrotoluene as internal standard, see Experimental section).73 It should be emphasized that this transformation on isatine opens up new routes for the synthesis of a plethora of interesting oxindole alkaloids related molecules.74
Silver catalysed domino Henry reaction/cycloisomerisation sequence
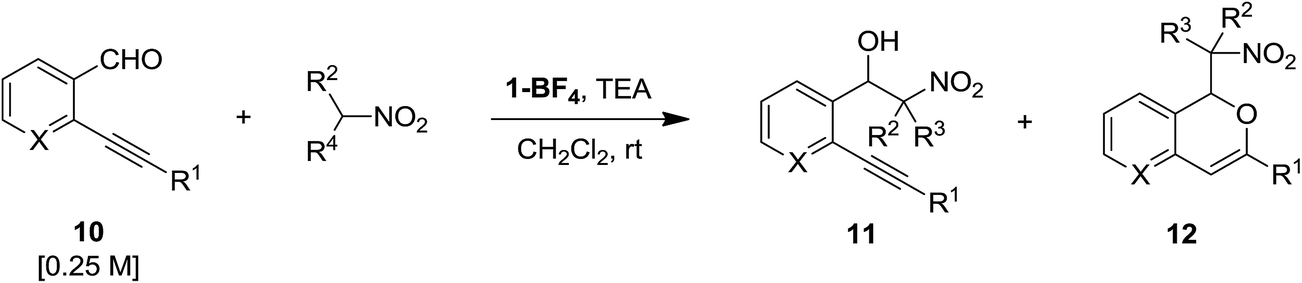
As mentioned above, we have recently reported that [silver(I)(Pc-L)] complexes are competent catalysts for the regioselective synthesis of 3-substituted-1-alkoxyisochromenes starting from 2-alkynylbenzaldehydes in the presence of alcohols as nucleophiles.61 Having seen their ability in promoting the Henry reaction, we were thus intrigued to see if it was possible to combine in a single domino sequence the Henry reaction of 2-alkynylbenzaldehydes and the cycloisomerisation to yield isochromenes 12. A two-step process that take advantage of a Cu(II) catalysed Henry reaction followed by an Au(I) mediated cycloisomerisation75 has recently been reported by Y. Gong and co-workers (Scheme 5).39 | ||
| Scheme 5 Alternative domino silver(I) catalysed synthesis of 1-isochromenes. | ||
The reaction between 2-[(4-methoxyphenyl)-ethynyl]benzaldehyde 10a and nitromethane was selected as model reaction for the optimization of reaction conditions. Firstly, we compared the activity copper(I) catalyst 2 and silver(I) catalyst 1-OTf (10 mol%) in dichloromethane in the presence of 5 equiv. of nitromethane and 10 mol% of TEA (Table 4, entries 1 and 2). This preliminary test interestingly revealed that while the copper complex 2 gave selectively the Henry reaction product 11a in 56% yield, the silver complex 1-OTf was able to promote the domino sequence, although with low selectivity, yielding the desired 1-isochromene 12a and the nitroalcohol 11a in equal amount, beside a 12% of unreacted aldehyde 10a and a complex mixture of unidentified by-products. An increase of the reaction temperature resulted in lower yields and selectivity, with an increase of by-products formation (Table 4, entries 3 and 4). The addition of an excess of TEA resulted in the selective formation of the Henry product in 45% yield, but was detrimental for the formation of the desired product 12a (Table 4, entry 5). As already stated above, while the Henry product can be formed also in the absence of the silver catalyst (although in poor yields), the presence of the metal is essential for the formation of the isochromene 12. In fact, in the presence of 1 equiv. of TEA and without the metal catalyst, the nitroalcohol 11a was selectively obtained in 77% yield (Table 4, entry 6).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base (equiv.) | MeNO2 (equiv.) | Solvent | t (h) | T (°C) | Yield 11ab (%) | Yield 12ab (%) | Rec. 10a(%) |
| a Reactions were performed with [Ag(I)] (2.5 × 10−2 mmol) in the solvent (1.25 mL) at a catalyst/aldehyde ratio of 1:10.b Yields based on initial 10a calculated via 1H NMR using 2,4-dinitrotoluene (DNT) as internal standard; isolated yields in brackets. Under these conditions, unreacted starting aldehyde did not always account for the rest of the reaction mass balance. In some cases, unidentified by-products derived from competitive side reactions were detected.c TEA (2.5 × 10−1 mmol)/aldehyde/nitromethane ratio of 1:1:5.d Reaction performed with freshly distilled nitromethane.e In the presence of molecular sieves (4 Å). |
|||||||||
| 1 | 2 | TEA (1) | 50 | CH2Cl2 | 20 | rt | (56) | — | (35) |
| 2 | 1-OTf | TEA (1) | 50 | CH2Cl2 | 20 | rt | (17) | (17) | (12) |
| 3 | 1-OTf | TEA (1) | 50 | CH2Cl2 | 30 | 40 | 12 | 6 | 7 |
| 4 | 1-OTf | TEA (1) | 50 | CH2Cl2 | 30 | 60 | 11 | 4 | 4 |
| 5 | 1-OTf | TEA (5) | 50 | CH2Cl2 | 22 | rt | 45 (39) | — | 11 (10) |
| 6c | — | TEAc | c | CH2Cl2 | 22 | rt | 77 | — | 10 |
| 7 | 1-OTf | TEA (1) | 50 | Tol | 20 | 30 | 30 | 8 | 11 |
| 8 | 1-OTf | TEA (1) | 50 | DMF | 24 | 30 | 53 | — | 6 |
| 9 | 1-OTf | TEA (1) | 50 | THF | 22 | rt | 46 | 2 | 21 |
| 10 | 1-OTf | TEA (1) | 11 | CH2Cl2 | 22 | rt | 3 | — | 5 |
| 11 | 1-OTf | TEA (1) | — | CH3NO2 | 20 | rt | 39 | 29 | — |
| 12 | AgOTf | TEA (1) | — | CH3NO2 | 20 | rt | — | 15 | 15 |
| 13d | 1-OTf | TEA (1) | 500 | CH2Cl2 | 22 | rt | 18 | 24 | — |
| 14d | 1-OTf | TEA (1) | 500 | CH2Cl2 | 22 | rt | 25 | 33 | — |
| 15d | 1-NTf2 | TEA (1) | 500 | CH2Cl2 | 22 | rt | 16 | 29 | 5 |
| 16d | 1-BF4 | TEA (1) | 500 | CH2Cl2 | 22 | rt | 21 (18) | 33 (30) | 10 (9) |
| 17d,e | 1-BF4 | TEA (1) | 500 | CH2Cl2 | 22 | rt | 60 (57) | 19 (17) | — |
| 18d,e | 1-BF4 | TEA (1) | 500 | CH3CN | 22 | rt | 96 | — | — |
| 19d | 1-BF4 | DIPEA (1) | 500 | CH2Cl2 | 22 | rt | 36 | 27 | — |
| 20d | 1-BF4 | DMAP (1) | 500 | CH2Cl2 | 22 | rt | 50 | — | 4 |
| 21d | 1-OTf | DBU (1) | 500 | CH2Cl2 | 22 | rt | 11 | 4 | 16 |
| 22d | 1-BF4 | NaHCO3 (1) | 500 | CH2Cl2 | 22 | rt | 5 | 1 | 65 |
| 23d | 1-OTf | Cs2CO3 (1) | 500 | CH2Cl2 | 22 | rt | 72 | — | 2 |
| 24d | 1-NTf2 | K2CO3 (1) | 500 | CH2Cl2 | 22 | rt | 78 | — | 2 |
| 25e | 8b-BF4 | TEA (1) | 500 | CH2Cl2 | 22 | rt | 57 | 15 | 8 |
| 26e | 8d | — | 500 | CH2Cl2 | 22 | rt | 60 | 15 | 13 |

The screening of solvent effect (Table 4, entries 7–9) revealed that polar aprotic solvents favour the formation of the nitroalcohol 11a. The reduction of the equivalent of nitromethane was detrimental to the reaction outcome (Table 4, entry 10), whereas, when CH3NO2 was employed as a reagent/solvent, the formation of 11a (39%) was accompanied by the formation of a discrete amount of isochromene 12a (29%, Table 4, entry 11), suggesting that an excess of nitroalkane is able to promote both isochromene and nitroalcohol formation. Interestingly, when simple AgOTf salt was used no trace of Henry product 11a was observed, and only a little amount of 12a was obtained (Table 4, entry 12).
Thus, a 50 fold excess of the nitromethane (with respect to aldehyde 10a) was used and this seems to favour the formation of the isochromene product, especially when freshly distilled nitromethane was used (Table 4, entries 13 and 14).
Among the counter anion tested, BF4− and OTf− displayed very close results, while NTf2− led to slightly lower overall yields (Table 4, entries 14–16). It is interesting to note that the presence of 4 Å molecular sieves as water scavenger seems to speed up the formation of the Henry product to the detriment of isochromene (Table 4, entry 17), and this selectivity became specificity when the reaction was performed in acetonitrile (Table 4, entry 18).
We tried to improve the formation of the isochromene 12a by using bases with different pKb, ranging from organic to inorganic ones (Table 4 entries 19–24), but the more acceptable results remained those obtained with TEA or with the more sterically demanding diisopropylethyl amine (DiPEA) (Table 4, entry 19).
Finally, the use of the catalytic systems previously selected for the Henry approach, confirm their tendency to promote preferentially the formation of the corresponding nitroalcohol (Table 4, entries 25 and 26), also in the absence of the additional base (Table 4, entry 26).
With the aim to explain the behaviour observed, we made an additional experiment. Isolated 11a was reacted in toluene at rt in the presence of 10 mol% of the silver(I) complex 1-OTf. After 24 h no reaction occurred and the TLC analysis showed the presence of unreacted 11a. Upon addition of 10 mol% of TEA the mixture was reacted for additional 24 h at rt, then the crude was analysed by 1H NMR which reveals the presence of unreacted 11a (44%) along with traces of 2-[(4-methoxyphenyl)-ethynyl]benzaldehyde 10a (≈5%), isochromene 11a (≈5%) and some unidentified by-products. This results suggest that the formation of the nitroalcohol 11a (Path B in Scheme 6) and the cascade synthesis of isochromene 12a (Path A in Scheme 6) are probably alternative and competitive pathways. According to reported metal catalysed domino synthesis of isochromenes in the presence of nucleophiles,61,76 12a is most likely formed by nucleophilic attack of the nitronate anion on a preformed isochromenilium ion intermediate I (Path A in Scheme 6), while a subsequent silver catalysed cycloisomerisation of the nitroalcohol 11a (Path C) seems to be most unlikely.77 Endorsing this hypothesis, under our reaction conditions we never isolated or detected in the reactions crude the alternative isobenzofuran isomers obtained by Gong and co-workers39 (see Scheme 5).
 | ||
| Scheme 6 Alternative/competitive pathways in the reaction of 2-alkynylarylalehydes and nitroalkanes. | ||
Up to now, any attempt to obtain in an exclusive fashion the desired isochromene 12a meet with failure, thus we decided to briefly investigate the substrate effect under the best reaction condition achieved for the synthesis of isochromenes (Table 4, entry 16). The results are reported in Table 5.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | SM | R1 | X | R2 | R3 | t (h) | Yield 11b (%) | Yield 12b (%) | Rec. SM (%) |
| a Reactions were performed with [Ag(I)] (2.5 × 10−2 mmol) in CH2Cl2 (1.25 mL) at a catalyst/TEA/aldehyde/nitromethane ratio of 1:1:10:500.b Isolated yields based on initial alkynylbenzaldehyde 10. Under these conditions, unreacted starting aldehyde did not always account for the rest of the reaction mass balance. In some cases, unidentified by-products derived from competitive side reactions were detected.c Mixture of two diastereoisomers in 70:30 ratio.d Mixture of two diastereoisomers in 75:25 ratio calculated on the 1H NMR.e In this case, the isocumarine 13 (29%) was recovered as major by-product.f In the presence of molecular sieves (4 Å). |
|||||||||
| 1 | 10a | p-MeO-Ph | CH | H | H | 22 | 11a 18 | 12a 30 | 9 |
| 2 | Me | H | 22 | 11b + 11b′ 70c | 12b + 12b′ 7d | 21 | |||
| 3 | Me | Me | 22 | — | — | 42e | |||
| 4 | 10b | p-Me-Ph | CH | H | H | 22 | 11c 32 | 12c 31 | 5 |
| 5f | H | H | 22 | 11c 60 | 12c 5 | 9 | |||
| 6 | 10c | p-CF3-Ph | CH | H | H | 22 | 11d 45 | 12d traces | 14 |
| 7 | 10d | Me3Si | CH | H | H | 22 | 11e 48 | — | 45 |
| 8f | H | H | 22 | 11e 83 | — | 15 | |||
| 9 | 10e | n-Pr | CH | H | H | 22 | 11f 15 | 12f 54 | — |
| 10 | 10f | n-Pr | N | H | H | 1 | 11g 52 | — | — |
| 11 | 10g | p-MeO-Ph | N | H | H | 22 | 11h 68 | — | 15 |
| 12f | H | H | 22 | 11h 78 | — | — | |||
All tested o-alkynylarylaldehydes 10 were readily obtained in moderate to excellent yields by PdCl2(PPh3)2 catalysed Sonogashira coupling reactions78 starting from commercially available 2-bromo(hetero)arylaldehydes and terminal acetylenes (see Experimental section for details).
Compared to the model reaction (Table 5, entry 1) when nitroethane was used under standard conditions we observed the formation of only a small amount of distereoisomeric isochromenes 12b and 12′b (not separated), while the Henry products 11b and 11′b were recovered in 70:30 diastereoisomeric ratio in 70% overall yield (Table 5, entry 2). On the other hand, the reaction with bulkier 2-nitropropane gave the 3-(4-methoxyphenyl)isocumarine 13 (29%) as main reaction by-product, beside a huge amount of unreacted starting material (42%) (Table 5, entry 3). The formation of 13 probably derived from the oxidation of the aldehyde 10a to a carboxylic acid, which was transformed by the alkaline reaction condition in a carboxylate nucleophile able to make an intramolecular attack to the silver(I) activated triple bond79 (Scheme 7).
 | ||
| Scheme 7 Plausible path for the formation of isocumarine 13. | ||
The presence of a neutral aryl substituent on the alkynylbenzaldehyde 10b gave an almost equal amount of nitroalcohol 11c, and isochromene 12c, (Table 5, entry 4). As already observed (Table 4, entries 16 and 17), the presence of traces of water seems to be important to promote the formation of the isochromene products 12; when the reaction of 10b was performed under strictly anhydrous conditions in the presence of 4 Å molecular sieves, the yield of 12c fall down while the yield of 11c doubled (Table 5, compare entries 4 and 5). The presence of EWG on the aryl moiety was not suitable for the formation of the isochromene 12d, and the corresponding nitroalcohol 11d was the main product obtained (Table 5, entry 6). In addition, trimethylsilyl substituted alkynylbenzaldehyde 10d, gave exclusively the nitroaldol product 11e (Table 5, entry 7), and when the reaction was conducted in the presence of a water scavenger, the yield of 11e skyrocket to 83% (Table 5, entry 8). Alkyl substitution on the triple bond was the only substitution that lead a good selectivity in favour of the cyclic product (Table 5, entry 9). More basic and coordinating nicotinaldehyde derivatives 10g,h yielded exclusively to the Henry products 10h and 10i (Table 5, entries 10 and 11). In the latter cases, the selectivity in favour of the Henry product could be explained by a speeding up of the nucleophilic attack of the nitronate anion to the aldehyde, promoted by the presence of an additional basic nitrogen on the pyridine moiety and by the EW activating effect of the electron-poor pyridine on the proximate aldehyde group. Also in this case, the presence of molecular sieves resulted in higher yields of nitroalcohol 11i (Table 5, entry 12).
Conclusions
The [Ag(I)(Pc-L)] complexes employed in the present study are suitable catalysts for the Henry reaction starting from various electron-poor aromatic aldehydes or other activated aldehydes such as furfural. Isatine can be converted into the corresponding nitroalcohol in excellent yields. Results obtained compares well with the state of the art and, at least compared to results obtained by us employing related copper(I) Pc-L complexes,22 better yields in shorter reaction times could be obtained. Advantages of the silver catalysed nitro–aldol reaction are: (i) mild reaction conditions, (ii) good to excellent reaction yields, (iii) cleanness of the reaction and (iv) total selectivity in nitroalcohol formation. All the silver(I) complexes reported are stable and can be handled in open-air atmosphere. Cs2CO3 demonstrated to be the base co-catalyst of choice in terms of reaction yields and reduced reaction times. Moreover, a careful design of the ligand has led to the synthesis of complex 8d, which contains a suitable basic functionality embedded into the ligand pendant, thus providing a bifunctional catalyst. A major advantage is represented by the straightforward synthesis of ligand 7d that is obtained in good overall yield starting from cheap starting materials. Further effort in our research group will be devoted to the structural modification of the ligand in order to control the enantioselectivity of this useful transformation.Based on our experience in [Ag(I)(Pc-L)] catalysed domino addition/cycloisomerisation reaction of 2-alkynylbenzaldehydes and nucleophiles, we tried to find a new entry to isochromenes by coupling the Henry reaction and the cycloisomerisation in a single domino process. We have demonstrated that even though with strong limitations, this cascade reaction can occur and that the mechanism involves a preliminary metal catalysed cycloisomerisation of the 2-alkynylbenzaldehyde to form an isochromenilium intermediate, followed by the attack from the nucleophilic nitronate anion. A catalytic amount of base is necessary to generate enough nitronate anion in the reaction medium. Interestingly, this pathway seems to be alternative and competitive with the formation of the nitroalcohol via the Ag catalysed Henry reaction. Since in our reaction conditions the Henry reaction is in general faster, this results in a non-selective process, yielding a mixture of nitroalcohols 11 and isochromenes 12. While is still possible to direct the reaction towards the nitroalcohol formation, a full selectivity toward the isochromene product has not yet been obtained.
Experimental procedures
General experimental details
All of the reactions that involved the use of reagents sensitive to oxygen or hydrolysis were carried out under an inert atmosphere. The glassware was previously dried in an oven at 110 °C and was set with cycles of vacuum and nitrogen. Also syringes, used to transfer reagents and solvents, were previously set under a nitrogen atmosphere. The syntheses of the silver complexes were carried out under a nitrogen atmosphere by employing standard Schlenk techniques. All chemicals and solvents were commercially available and were used after distillation or treatment with drying agents. The chromatographic column separations were performed by a flash technique, using silica gel (pore size 60 Å, particle size 230–400 mesh, Merck grade 9385). For TLC, silica was used on TLC Alu foils with fluorescent indicator (254 nm) and the detection was performed by irradiation with UV light (λ = 254 nm or 366 nm). 1H NMR analyses were performed with 200, 300, 400 or 600 MHz spectrometers at room temperature. The coupling constants (J) are expressed in hertz (Hz), and the chemical shifts (δ) in ppm. 13C NMR analyses were performed with the same instruments at 75.5, 100 or 150 MHz, and attached proton test (APT) sequence was used to distinguish the methine and methyl carbon signals from those arising from methylene and quaternary carbon atoms. All 13C NMR spectra were recorded with complete proton decoupling. The 1H NMR signals of the ligand described in the following have been attributed by correlation spectroscopy (COSY) and nuclear Overhauser effect spectroscopy (NOESY) techniques. Assignments of the resonance in 13C NMR were made using the APT pulse sequence and heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) techniques. Low resolution MS spectra were recorded with instruments equipped with electron ionization (EI), ESI/ion trap (using a syringe pump device to directly inject sample solutions), or fast atom bombardment (FAB) (for Pc-L and metal complexes) sources. The values are expressed as mass-charge ratio and the relative intensities of the most significant peaks are shown in brackets. UV-vis spectra of the ligand and its silver complexes were recorded in CH2Cl2. Elemental analyses were recorded in the analytical laboratories of Università degli Studi di Milano. Optical rotations were measured on a Perkin Elmer instruments model 343 plus; [α]D values are given in 10−1 deg cm2 g−1. Silver complex 1,60 N-tosyl-aziridine and copper complex 250 and 2,6-pyridinedimethanol 2,6-dimesylate61 were synthetized as previously reported.Synthesis of β-alanine methyl ester hydrochloride
TMS-Cl (8.7 mL, 68.6 mmol) was added dropwise to β-Ala (3.06 g, 34.3 mmol). Then MeOH (27.0 mL) was added slowly and the resulting solution was stirred for 24 h at rt. The solvent was evaporated to dryness leading to (2) (4.8 g, >99%) as a white solid. 1H NMR (300 MHz, D2O, δ) 3.81 (s, 3H, OCH3), 3.36 (t, J = 6.4 Hz, 2H, CH2), 2.89 (t, J = 6.4 Hz, 2H, CH2). The spectral data are consistent with those previously reported.71Synthesis of N(α)-Cbz-lysine methyl ester hydrochloride
TMS-Cl (0.9 mL, 7.0 mmol) was added dropwise to N(α)-Cbz-lysine (0.98 g, 3.0 mmol). Then MeOH (10.0 mL) was added slowly and the resulting solution was stirred for 24 h at rt. The solvent was evaporated to dryness leading to (3) (1.18 g, quantitative yield) as a colorless oil. 1H NMR (300 MHz, D2O, δ) 7.62–7.21 (m, 7H, Har and NH2), 5.20 (s, 2H, CH2), 4.28 (m, 1H, CH), 3.80 (s, 3H, OCH3), 3.01 (m, 2H, CH2), 2.39 (s, 1H, NH), 2.06–1.63 (m, 4H, CH2), 1.49 (m, 2H, CH2). The spectral data are consistent with those previously reported.80Synthesis of 5a and 6a
β-Alanine methyl ester hydrochloride (0.377 g, 2.7 mmol) was suspended in CH3CN (10.0 mL). TEA (0.45 mL, 3.22 mmol) was added, the mixture was warmed at 60 °C and half of a solution of N-tosyl aziridine (1.33 g, 6.74 mmol in 10.0 mL of toluene) was added dropwise in 10 minutes. After one hour was added dropwise the second half of the solution and the mixture was left to react for 6 h at reflux temperature. The reaction solvent was evaporated to dryness and the crude was purified by flash chromatography (SiO2, AcOEt/n-hexane 6:4 gradient to AcOEt/MeOH 20%) yielding to 6a (378 mg, yield 47%) and 5a (402 mg, yield 30%).
Synthesis of 5a from 6a
A solution of N-tosyl aziridine (278 mg, 1.45 mmol in 5.0 mL of toluene) was added dropwise to a solution of compound 6a (364 mg, 1.21 mmol in 5.0 mL of toluene) at 60 °C. The mixture was left to react for 8 h at reflux temperature. The solvent was evaporated to dryness and the crude was purified by flash chromatography (SiO2, AcOEt/n-hexane 7:3 gradient to AcOEt/n-hexane 9:1) yielding to 5a (503 mg, yield 84%) as a white foam. The spectral data are consistent with those reported above.
Synthesis of 5b and 6b
N(α)-Cbz-lysine methyl ester hydrochloride (0.608 g, 1.8 mmol) was suspended in CH3CN (6.0 mL). TEA (0.37 mL, 2.7 mmol) was added, the mixture was warmed at 60 °C and half of a solution of N-tosyl aziridine (1.05 g, 5.35 mmol in 16.0 mL of toluene) was added dropwise in 10 min. After one hour was added dropwise the second half of the solution and the mixture was left to react for 6 h at reflux temperature. The reaction solvent was evaporated to dryness and the crude was purified by flash chromatography (SiO2, AcOEt/n-hexane 6:4 gradient to AcOEt/MeOH 20%) yielding to (6b) (355 mg, yield 40%) and (5b) (552 mg, yield 45%).
Synthesis of 5b from 6b
A solution of N-tosyl aziridine (168 mg, 0.85 mmol in 4.0 mL of toluene) was added dropwise to a solution of compound 6b (350 mg, 0.71 mmol in 4.0 mL of toluene) at 60 °C. The mixture was left to react for 8 h at reflux temperature. The solvent was evaporated to dryness and the crude was purified by flash chromatography (SiO2, AcOEt/n-hexane 7:3 gradient to AcOEt/n-hexane 9:1) yielding to 5b (442 mg, yield 90%) as a white foam. The spectral data are consistent with those reported above.
Synthesis of 7a
Compound 5a (201 mg, 0.40 mmol) was dissolved in anhydrous CH3CN (12.0 mL). 2,6-Pyridinedimethanol 2,6-dimesylate (120 mg, 0.40 mmol) and K2CO3 (165 mg, 1.2) were added and the resulting mixture was left to react for 6 h at reflux temperature. The solvent was evaporated to dryness and the crude was suspended in 20 mL of water and extracted with AcOEt (3 × 10.0 mL). The organic phase was washed with brine (2 × 15.0 mL), treated with Na2SO4 and filtered. The solvent was evaporated to dryness yielding to 7a (232 mg, yield 97%) as a light yellow solid. 1H NMR (400 MHz, CDCl3, δ) 7.77–7.69 (m, 5H, Har), 7.37–7.29 (m, 6H, Har), 4.32 (s, 4H, CH2), 3.64 (s, 3H, OCH3), 3.10 (m, 4H, CH2), 2.65 (m, 2H, CH2), 2.44 (s, 6H, CH3), 2.35–2.24 (m, 6H, CH2). 13C-NMR (100 MHz, CDCl3, δ) 172.8 (CO), 155.2 (Car), 143.7 (Car), 138.9 (CHar), 130.0 (CHar), 127.3 (CHar), 124.2 (CHar), 54.7 (CH2), 51.7 (CH2), 51.3 (OCH3), 51.1 (CH2), 45.2 (CH2), 34.0 (CH2), 22.9 (CH2), 21.7 (CH3). MS (ESI): m/z (%) = 601.3 (100) [MH]+. Anal. calcd for C29H36N4O6S2: C, 57.98; H, 6.04; N, 9.33. Found: C, 57.62; H, 5.92; N, 9.12. UV/vis (5.2 × 10−5 mol L−1, CH2Cl2 in 1 cm cuvettes): λmax [nm] = 234 nm. IR (ATR): 2949 cm−1 (w), 1733 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1157 cm−1.
O), 1157 cm−1.
Synthesis of 7b
Compound 5b (280 mg, 0.40 mmol) was dissolved in anhydrous CH3CN (14.0 mL). 2,6-Pyridinedimethanol 2,6-dimesylate (123 mg, 0.42 mmol) and K2CO3 (184 mg, 1.3 mmol) were added and the resulting mixture was left to react for 6 h at reflux temperature. The solvent was evaporated to dryness and the crude was suspended in 25 mL of water and extracted with AcOEt (3 × 15.0 mL). The organic phase was washed with brine (2 × 20.0 mL), treated with Na2SO4 and filtered. The solvent was evaporated to dryness yielding to 7b (318 mg, 97%) as a light yellow solid. 1H NMR (300 MHz, CDCl3, δ) 7.73–7.69 (m, 5H, Har), 7.38–7.27 (m, 11H, Har), 5.29 (m, 1H, NH-Cbz), 5.11 (s, 2H, CH2), 4.39–4.28 (m, 5H, CH and CH2), 3.74 (s, 3H, OCH3), 3.10 (m, 4H, CH2), 2.44 (s, 6H, CH3), 2.32–2.20 (m, 6H, CH2), 1.79 (m, 1H, CH2), 1.63 (m, 1H, CH2), 1.36–1.16 (m, 4H, CH2). 13C-NMR (75 MHz, CDCl3, δ) 173.1 (CO), 155.1 (C), 143.7 (Car), 138.9 (CHar), 136.2 (Car), 130.0 (CHar), 128.7 (CHar), 128.3 (CHar), 127.3 (CHar), 124.2 (CHar), 67.1 (CH2), 55.2 (CH2), 54.6 (CH2), 54.0 (CH2), 52.5 (OCH3), 51.8 (CH2), 44.8 (CH2), 32.6 (CH2), 27.78 (CH2), 22.8 (CH2), 21.7 (CH3). MS (ESI): m/z (%) = 792.4 (100) [MH]+. Anal. calcd for C40H49N5O8S2: C, 60.66; H, 6.24; N, 8.84. Found: C, 60.28; H, 5.96; N, 8.60. [α]20D = 3.82° (c 1.00 in CHCl3). UV/vis (5.0 × 10−5 mol L−1, CH2Cl2 in 1 cm cuvettes): λmax [nm] = 234 nm. IR (ATR): 3337 cm−1 (NH), 2932 (w), 1750 cm−1 (νCO), 1152 cm−1.
Synthesis of 7c
Under H2 atmosphere, compound 7b (700 mg, 0.88 mmol) was dissolved in MeOH (15.0 mL). Pd/C (70 mg) was added and the resulting suspension was left to react for 24 h at rt. The suspension was filtered on celite layer and celite was washed with MeOH (3 × 5.0 mL). The solvent was evaporated to dryness yielding to 7c (579 mg, 99%) as a pale yellow foam. 1H NMR (300 MHz, CD3OD, δ) 7.84 (d, J = 8.3 Hz, 4H, Har), 7.75 (pst, J = 7.7 Hz, 1H, Har), 7.75 (d, J = 8.3 Hz, 4H, Har), 7.26 (d, J = 7.7 Hz, 2H, Har), 4.59 (m, 2H, CH2), 4.32 (m, 2H, CH2), 4.15 (m, 1H, CH) overlapping with 4.7–4.13 (m, 2H, CH2), 3.88 (s, 3H, OCH3), 3.75 (m, 2H, CH2), 3.70–3.48 (m, 4H, CH2), 3.16 (m, 2H, CH2), 2.45 (s, 6H, CH3), 2.14 (m, 2H, CH2), 1.97 (m, 2H, CH2), 1.73–1.59 (m, 2H, CH2). 13C-NMR (75 MHz, CDCl3, δ) 171.2 (CO), 159.3 (Car), 146.5 (Car), 140.3 (CHar), 134.7 (Car), 131.4 (CHar), 129.2 (CHar), 122.6 (CHar), 53.8 (OCH3), 53.7 (CH), 53.4 (CH2), 50.3 (CH2), 46.7 (CH2), 43.5 (CH2), 31.1 (CH2), 23.3 (CH2), 21.5 (CH3), 20.5 (CH2). MS (ESI): m/z (%) = 658.3 (100) [MH]+. Anal. calcd for C32H43N5O6S2: C, 58.42; H, 6.59; N, 10.65. Found: C, 58.54; H, 6.50; N, 10.71.Synthesis of 7d
Compound 7c (579 mg, 0.88 mmol) was dissolved in AcOH (1.5 mL). NaCNBH3 (166 mg, 2.65 mmol) was added and the mixture was left to stir 10 min at rt. Acetaldehyde (1.1 mL, 19.36 mmol) was added in 72 h in small amounts (4 eq. every 12 h). The solvent was evaporated to dryness, then brine (10.0 mL) was added and the solution was extracted with DCM (5 × 10.0 mL). The organic phase was treated with Na2SO4 and filtered. The solvent was evaporated to dryness yielding to 7d (376 mg, yield 60%) as a light yellow foam. 1H NMR (400 MHz, CDCl3, δ) 7.72 (d, J = 8.2 Hz, 5H, HAr), 7.32 (t, J = 8.5 Hz, 6H, HAr), 4.34 (s, 4H, CH2), 3.67 (s, 3H, OCH3), 3.31 (t, J = 7.4 Hz, 1H, CH), 3.15–3.03 (m, 4H, CH2), 2.70 (dq, J = 14.5, 7.2 Hz, 2H, CH2), 2.51–2.40 (m, 8H, CH2 + CH3), 2.27 (dd, J = 16.6, 9.9 Hz, 6H, CH2), 1.60 (ddd, J = 15.6, 14.6, 9.2 Hz, 2H, CH2), 1.31 (m, 2H, CH2), 1.02 (t, J = 7.1 Hz, 6H, CH3). 13C NMR (100 MHz, CDCl3, δ) 174.2 (CO), 155.1 (CAr), 143.6 (CAr), 138.9 (CHAr), 136.2 (CAr), 130.0 (CHAr), 127.3 (CHAr), 124.2 (CHAr), 110.7 (CH), 63.2 (CH), 55.5 (CH2), 54.8 (CH2), 51.7 (CH2), 51.1 (CH), 44.7 (CH2), 29.9 (CH2), 28.3 (CH2), 24.1 (CH2), 21.7 (CH3), 14.1 (CH3). MS (ESI): m/z (%) = 712.3 (100) [MH]+. Anal. calcd for C36H51N5O6S2: C, 60.56; H, 7.20; N, 9.81. Found: C, 60.22; H, 7.12; N, 10.02.Reaction of ligand 7c with (R)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride
(R)-(−)-α-Methoxy-α-(trifluoromethyl)phenylacetyl chloride (17 mg, 0.067 mmol) was added to a solution of ligand 7c (40 mg, 0.061 mmol) in CD3COCD3 (1.0 mL), in the presence of pyridine (14 mg, 0.18 mmol). The solution was stirred for 30 min, and then it was transferred into a NMR test tube. 19F NMR (282 MHz, CDCl3, δ) −71.24.Reaction of ligand 7c with α-methoxyphenylacetic acid
To a solution of ligand 7c (21 mg, 0.032 mmol) in CDCl3 (1.0 mL) were added, in this order, (S)-(+) or (R)-(−)-α-methoxyphenylacetic acid (5 mg, 0.032 mmol), N,N′-dicyclohexylcarbodiimide (13 mg, 0.065 mmol) and 4-dimethylaminopyridine (3 mg, 0.022 mmol). The solution was stirred for 30 min, and then it was transferred into a NMR test tube.General procedure for the synthesis of Ag(I) complexes
Under N2, the ligand (0.39 mmol) was dissolved in anhydrous C2H4Cl2 (10.0 mL), Ag(I) was added (0.39 mmol) as AgBF4 or AgOTf and the mixture was left to react for 1 h at rt. The suspension was filtered under N2, the solvent was concentrated to ∼1.0 mL and anhydrous n-hexane (10.0 mL) was added. The mixture was left for 10 min at rt, and then the solvent was evaporated to dryness. n-Hexane (10.0 mL) was added and the suspension was left under stirring over night at rt, then was opened at air and filtered yielding to the corresponding Ag(I) complex as a white powder.O), 1155 cm−1.O), 1161 cm−1.O), 1156 cm−1.O), 1160 cm−1.General procedure for the Henry reaction
The reactions were performed under a nitrogen atmosphere. The catalyst (0.032 mmoles) was dissolved in dry CH2Cl2 (5.0 mL) at rt in a Schlenk flask equipped with a stirring bar (were indicated molecular sieves-4 Å were added, see Tables 1–3 captions). Reactants were then added in the following order: the proper aldehyde 3 (0.32 mmoles), the nitroalkane (1.6 mmoles), and finally the selected base (0.032 mmoles). The resulting mixture was stirred at 30 °C for 12 h, extracted three times with brine. The organic layer was dried over sodium sulphate, and the solvent was evaporated under reduced pressure. The reaction crude was purified by flash column chromatography over a silica gel column with gradients of n-hexane/ethyl acetate as eluent. Products 4h, 4n and 4p (yields below 15%) were not isolated.General procedure for the synthesis of 2-alkynylbenzaldehydes 10a–e and 2-alkynylnicotinaldehydes 10f–g
To a solution of 2-bromobenzaldehyde or 2-bromonicotinaldehyde (3.24 mmol) in dry triethylamine (97.2 mmol), the appropriate alkyne (3.89 mmol) and trans-dichlorobis(triphenylphosphine)palladium(II) (2 mol%) were added, under a nitrogen atmosphere. The reaction was stirred at rt for 10 min, and then CuI (1 mol%) was added. The reaction mixture was stirred at 50 °C until no more starting product was detectable by TLC analysis (eluent: n-hexane/ethyl acetate). The solvent was then evaporated under reduced pressure and the crude material was purified by flash chromatography over a silica gel column with gradients of n-hexane/ethyl acetate as eluent. 2-Alkynylbenzaldehydes 10a,61 b,61 c,81 d,61 and e,61 and 2-alkynylnicotinaldehydes f82 and g83 are known compounds. They were characterised by 1H-NMR and spectral data are in good agreement with literature values.General procedure for the reaction of 2-alkynylarylalehydes 10a–g with nitroalkanes
The reaction were performed in a 0.25 mmol scale under a nitrogen atmosphere. The catalyst 1-BF4 (0.025 mmol) was dissolved in dry CH2Cl2 (1.25 mL) in a Schlenk test tube equipped with a stirring bar. The mixture was stirred at rt for 10 min. The proper 2-alkynylarylalehyde 10 (0.25 mmol), nitroalkane (12.5 mmol) and triethylamine (0.025 mmol) were added to the stirred solution, in this order. In some cases (see Table 5), 50 mg of 4 Å molecular sieves were added. The mixture was stirred at rt for 1–22 h (for reaction times see Table 5). The reaction mixture was diluted with CH2Cl2 (20.0 mL) and the organic layer was washed twice with brine (2 × 20.0 mL). The organic layer was dried over sodium sulphate, and the solvent was evaporated under reduced pressure. The reaction crude was purified by flash column chromatography over a silica gel column with gradients of n-hexane/ethyl acetate as eluent.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :25 ratio). Yield 7% (6 mg); 1H NMR (300 MHz, CDCl3, δ) 7.64 (d, J = 9.0 Hz, 2H, HAr), 7.57 (d, J = 9.0 Hz, 2H, HAr), 7.36–7.11 (m, 4 + 4H, HAr), 6.94–6.89 (m, 2 + 2H, HAr), 6.39 (s, 1H, HAr), 6.30 (s, 1H, HAr), 5.90 (d, J = 5.9 Hz, 1H, –CH–O), 5.68 (d, J = 9.7 Hz, 1H, –CH–O), 5.09 (dq, J = 9.7, 6.8 Hz, 1H, –CH–NO2), 4.93 (dq, J = 6.7, 5.9 Hz, 1H, –CH–NO2), 3.85 (s, 3H, CH3–O), 3.83 (s, 3H, CH3–O), 1.66 (d, J = 6.7 Hz, 3H, CH3), 1.39 (d, J = 6.8 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3, δ) 160.9 (CAr), 160.8 (CAr), 151.4 (Cq), 151.0 (Cq), 131.4 (CAr), 131.1 (CAr), 129.8 (CHAr), 129.5 (CHAr), 127.1 (CHAr), 126.9 (CHAr), 126.6 (CHAr), 126.5 (CHAr), 126.4 (CHAr), 126.1 (CAr), 125.7 (CAr), 125.0 (CHAr), 124.53 (CHAr), 124.48 (CAr), 124.42 (CHAr), 114.2 (CHAr), 114.1 (CHAr), 98.7 (CHAr), 98.6 (CHAr), 85.7 (CH), 82.3 (CH), 79.5 (CH), 78.8 (CH), 55.56 (CH3–O), 55.54 (CH3–O), 16.22 (CH3), 13.94 (CH3) (one quaternary carbon of the minority compound 12b′ obscured). ESI-MS m/z (%): 312 [M + H]+ (100).
:25 ratio). Yield 7% (6 mg); 1H NMR (300 MHz, CDCl3, δ) 7.64 (d, J = 9.0 Hz, 2H, HAr), 7.57 (d, J = 9.0 Hz, 2H, HAr), 7.36–7.11 (m, 4 + 4H, HAr), 6.94–6.89 (m, 2 + 2H, HAr), 6.39 (s, 1H, HAr), 6.30 (s, 1H, HAr), 5.90 (d, J = 5.9 Hz, 1H, –CH–O), 5.68 (d, J = 9.7 Hz, 1H, –CH–O), 5.09 (dq, J = 9.7, 6.8 Hz, 1H, –CH–NO2), 4.93 (dq, J = 6.7, 5.9 Hz, 1H, –CH–NO2), 3.85 (s, 3H, CH3–O), 3.83 (s, 3H, CH3–O), 1.66 (d, J = 6.7 Hz, 3H, CH3), 1.39 (d, J = 6.8 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3, δ) 160.9 (CAr), 160.8 (CAr), 151.4 (Cq), 151.0 (Cq), 131.4 (CAr), 131.1 (CAr), 129.8 (CHAr), 129.5 (CHAr), 127.1 (CHAr), 126.9 (CHAr), 126.6 (CHAr), 126.5 (CHAr), 126.4 (CHAr), 126.1 (CAr), 125.7 (CAr), 125.0 (CHAr), 124.53 (CHAr), 124.48 (CAr), 124.42 (CHAr), 114.2 (CHAr), 114.1 (CHAr), 98.7 (CHAr), 98.6 (CHAr), 85.7 (CH), 82.3 (CH), 79.5 (CH), 78.8 (CH), 55.56 (CH3–O), 55.54 (CH3–O), 16.22 (CH3), 13.94 (CH3) (one quaternary carbon of the minority compound 12b′ obscured). ESI-MS m/z (%): 312 [M + H]+ (100).Acknowledgements
Financial support from the Università degli Studi di Milano (Sviluppo Unimi Bando TRANSITION GRANT – HORIZON 2020 – Linea A1_B Progetto “Italia per l'Europa. Cod.: 18499” is gratefully acknowledged. A. C. and G. T. are grateful to Flavia Roncalli for her precious help in the synthesis of 7a and 7b.Notes and references
- C. Cassani, G. Bergonzini and C.-J. Wallentin, ACS Catal., 2016, 6, 1640–1648 CrossRef CAS.
- R. Narayan, K. Matcha and A. P. Antonchick, Chem.–Eur. J., 2015, 21, 14678–14693 CrossRef CAS PubMed.
- R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC.
- A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed.
- R. Ballini, S. Gabrielli and A. Palmieri, in Green Chemistry for Environmental Sustainability, ed. S. K. Sharma and A. Mudhoo, CRC Press, Boca Raton, 2011, pp. 53–78 Search PubMed.
- N. Ono, The Nitro Group in Organic Synthesis, John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- L. Henry, Compt. Rend., 1895, 120, 1265–1268 CAS.
- H. Sasai, in Comprehensive Organic Synthesis II, Elsevier, Amsterdam, 2nd edn, 2014, pp. 543–570 Search PubMed.
- R. Ballini, L. Barboni, F. Fringuelli, A. Palmieri, F. Pizzo and L. Vaccaro, Green Chem., 2007, 9, 823–838 RSC.
- F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS.
- E. Chinnaraja, R. Arunachalam, M. K. Choudhary, R. I. Kureshy and P. S. Subramanian, Appl. Organomet. Chem., 2016, 30, 95–101 CrossRef CAS.
- N. Q. Shixaliyev, A. M. Maharramov, A. V. Gurbanov, V. G. Nenajdenko, V. M. Muzalevskiy, K. T. Mahmudov and M. N. Kopylovich, Catal. Today, 2013, 217, 76–79 CrossRef CAS.
- C. Pettinari, F. Marchetti, A. Cerquetella, R. Pettinari, M. Monari, T. C. O. Mac Leod, L. Martins and A. J. L. Pombeiro, Organometallics, 2011, 30, 1616–1626 CrossRef CAS.
- M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS PubMed.
- C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2007, 2561–2574 CrossRef.
- X. Yu, B. Perez, Z. Zhang, R. Gao and Z. Guo, Green Chem., 2016, 18, 2753–2761 RSC.
- S. E. Milner, T. S. Moody and A. R. Maguire, Eur. J. Org. Chem., 2012, 2012, 3059–3067 CrossRef CAS.
- V. E. Collier, N. C. Ellebracht, G. I. Lindy, E. G. Moschetta and C. W. Jones, ACS Catal., 2016, 6, 460–468 CrossRef CAS.
- N. A. Brunelli and C. W. Jones, J. Catal., 2013, 308, 60–72 CrossRef CAS.
- Y. Alvarez-Casao, E. Marques-Lopez and R. P. Herrera, Symmetry, 2011, 3, 220 CrossRef CAS.
- F. Xu, L. Yan, C. Lei, H. Zhao and G. Li, Tetrahedron: Asymmetry, 2015, 26, 338–343 CrossRef CAS.
- B. Castano, T. Pedrazzini, M. Sisti, E. Gallo, F. Ragaini, N. Casati and A. Caselli, Appl. Organomet. Chem., 2011, 25, 824–829 CrossRef CAS.
- W. Jin, X. Li and B. Wan, J. Org. Chem., 2011, 76, 484–491 CrossRef CAS PubMed.
- Y. Qiong ji, G. Qi and Z. M. A. Judeh, Eur. J. Org. Chem., 2011, 2011, 4892–4898 CAS.
- M. L. Ingalsbe, J. D. St Denis, J. L. Gleason, G. P. Savage and R. Priefer, Synthesis, 2010, 2010, 98–102 CrossRef.
- H. Y. Kim and K. Oh, Org. Lett., 2009, 11, 5682–5685 CrossRef CAS PubMed.
- K. Y. Spangler and C. Wolf, Org. Lett., 2009, 11, 4724–4727 CrossRef CAS PubMed.
- T. Arai, R. Takashita, Y. Endo, M. Watanabe and A. Yanagisawa, J. Org. Chem., 2008, 73, 4903–4906 CrossRef CAS PubMed.
- B. Qin, X. Xiao, X. Liu, J. Huang, Y. Wen and X. Feng, J. Org. Chem., 2007, 72, 9323–9328 CrossRef CAS PubMed.
- Y. Xiong, F. Wang, X. Huang, Y. Wen and X. Feng, Chem.–Eur. J., 2007, 13, 829–833 CrossRef CAS PubMed.
- D. Scharnagel, A. Mueller, F. Prause, M. Eck, J. Goller, W. Milius and M. Breuning, Chem.–Eur. J., 2015, 21, 12488–12500 CrossRef CAS PubMed.
- L.-W. Tang, X. Dong, Z.-M. Zhou, Y.-Q. Liu, L. Dai and M. Zhang, RSC Adv., 2015, 5, 4758–4765 RSC.
- X. Wang, W. Zhao, G. Li, J. Wang, G. Liu, L. Liu, R. Zhao and M. Wang, Appl. Organomet. Chem., 2014, 28, 892–899 CrossRef CAS.
- Q. Dai, N. K. Rana and J. C.-G. Zhao, Org. Lett., 2013, 15, 2922–2925 CrossRef CAS PubMed.
- M. Solinas, B. Sechi, S. Baldino and G. Chelucci, J. Mol. Catal. A: Chem., 2013, 378, 206–212 CrossRef CAS.
- L. Yao, Y. Wei, P. Wang, W. He and S. Zhang, Tetrahedron, 2012, 68, 9119–9124 CrossRef CAS.
- G. Blay, V. Hernandez-Olmos and J. R. Pedro, Synlett, 2011, 1195–1211 CrossRef CAS.
- G. Blay, V. Hernández-Olmos and J. R. Pedro, Chem.–Eur. J., 2011, 17, 3768–3773 CrossRef CAS PubMed.
- D. Lu, Y. Zhou, Y. Li, S. Yan and Y. Gong, J. Org. Chem., 2011, 76, 8869–8878 CrossRef CAS PubMed.
- H.-Y. Zhang, L. Chen, H.-B. Song and G.-F. Zi, Inorg. Chim. Acta, 2011, 366, 320–336 CrossRef CAS.
- L. Cheng, J. Dong, J. You, G. Gao and J. Lan, Chem.–Eur. J., 2010, 16, 6761–6765 CrossRef CAS PubMed.
- M. Luo and B. Yan, Tetrahedron Lett., 2010, 51, 5577–5580 CrossRef CAS.
- V. J. Mayani, S. H. R. Abdi, R. I. Kureshy, N.-U. H. Khan, A. Das and H. C. Bajaj, J. Org. Chem., 2010, 75, 6191–6195 CrossRef CAS PubMed.
- A. Noole, K. Lippur, A. Metsala, M. Lopp and T. Kanger, J. Org. Chem., 2010, 75, 1313–1316 CrossRef CAS PubMed.
- D.-M. Du, S.-F. Lu, T. Fang and J. Xu, J. Org. Chem., 2005, 70, 3712–3715 CrossRef CAS PubMed.
- Y. Zhang, L. Xiang, Q. Wang, X.-F. Duan and G. Zi, Inorg. Chim. Acta, 2008, 361, 1246–1254 CrossRef CAS.
- J. Gao and A. E. Martell, Org. Biomol. Chem., 2003, 1, 2801–2806 CAS.
- M. Rezaeivala and H. Keypour, Coord. Chem. Rev., 2014, 280, 203–253 CrossRef CAS.
- B. Castano, E. Gallo, D. J. Cole-Hamilton, V. Dal Santo, R. Psaro and A. Caselli, Green Chem., 2014, 16, 3202–3209 RSC.
- B. Castano, S. Guidone, E. Gallo, F. Ragaini, N. Casati, P. Macchi, M. Sisti and A. Caselli, Dalton Trans., 2013, 42, 2451–2462 RSC.
- B. Castano, P. Zardi, Y. C. Honemann, A. Galarneau, E. Gallo, R. Psaro, A. Caselli and V. Dal Santo, RSC Adv., 2013, 3, 22199–22205 RSC.
- A. Caselli, F. Cesana, E. Gallo, N. Casati, P. Macchi, M. Sisti, G. Celentano and S. Cenini, Dalton Trans., 2008, 4202–4205 RSC.
- V. K.-Y. Lo, A. O.-Y. Chan and C.-M. Che, Org. Biomol. Chem., 2015, 13, 6667–6680 CAS.
- G. Abbiati and E. Rossi, Beilstein J. Org. Chem., 2014, 10, 481–513 CrossRef PubMed.
- R. J. Scamp, J. W. Rigoli and J. M. Schomaker, Pure Appl. Chem., 2014, 86, 381–393 CrossRef CAS.
- M. Harmata, Silver in Organic Chemistry, John Wiley & Sons, Inc, Hoboken, New Jersey, 2010 Search PubMed.
- M. Alvarez-Corral, M. Munoz-Dorado and I. Rodriguez-Garcia, Chem. Rev., 2008, 108, 3174–3198 CrossRef CAS PubMed.
- M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132–3148 CrossRef CAS PubMed.
- Y. Yamamoto, Chem. Rev., 2008, 108, 3199–3222 CrossRef CAS PubMed.
- T. Pedrazzini, P. Pirovano, M. Dell'Acqua, F. Ragaini, P. Illiano, P. Macchi, G. Abbiati and A. Caselli, Eur. J. Inorg. Chem., 2015, 2015, 5089–5098 CrossRef CAS.
- M. Dell'Acqua, B. Castano, C. Cecchini, T. Pedrazzini, V. Pirovano, E. Rossi, A. Caselli and G. Abbiati, J. Org. Chem., 2014, 79, 3494–3505 CrossRef PubMed.
- M. Trose, M. Dell'Acqua, T. Pedrazzini, V. Pirovano, E. Gallo, E. Rossi, A. Caselli and G. Abbiati, J. Org. Chem., 2014, 79, 7311–7320 CrossRef CAS PubMed.
- K. Lang, J. Park and S. Hong, J. Org. Chem., 2010, 75, 6424–6435 CrossRef CAS PubMed.
- M. Dell'Acqua, V. Pirovano, G. Confalonieri, A. Arcadi, E. Rossi and G. Abbiati, Org. Biomol. Chem., 2014, 12, 8019–8030 Search PubMed.
- A. Arcadi, G. Abbiati and E. Rossi, J. Organomet. Chem., 2011, 696, 87–98 CrossRef CAS.
- M. Dell'Acqua, G. Abbiati, A. Arcadi and E. Rossi, Org. Biomol. Chem., 2011, 9, 7836–7848 Search PubMed.
- D. Facoetti, G. Abbiati, L. d'Avolio, L. Ackermann and E. Rossi, Synlett, 2009, 2009, 2273–2276 CrossRef.
- G. Abbiati, A. Casoni, V. Canevari, D. Nava and E. Rossi, Org. Lett., 2006, 8, 4839–4842 CrossRef CAS PubMed.
- G. Abbiati, A. Arcadi, A. Bellinazzi, E. Beccalli, E. Rossi and S. Zanzola, J. Org. Chem., 2005, 70, 4088–4095 CrossRef CAS PubMed.
- J. E. Richman and T. J. Atkins, J. Am. Chem. Soc., 1974, 96, 2268–2270 CrossRef CAS.
- J. Li and Y. Sha, Molecules, 2008, 13, 1111 CrossRef CAS PubMed.
- M. N. Elinson, A. I. Ilovaisky, V. M. Merkulova, F. Barba and B. Batanero, Tetrahedron, 2008, 64, 5915–5919 CrossRef CAS.
- W. R. Conn and H. G. Lindwall, J. Am. Chem. Soc., 1936, 58, 1236–1239 CrossRef CAS.
- L. Liu, S. Zhang, F. Xue, G. Lou, H. Zhang, S. Ma, W. Duan and W. Wang, Chem.–Eur. J., 2011, 17, 7791–7795 CrossRef CAS PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- (a) P. Belmont, in Silver in Organic Chemistry, ed. M. Harmata, John Wiley & Sons, Inc, Hoboken, New Jersey, 2010, ch. 5, pp. 143–165 Search PubMed. See also: (b) A. Stephen and K. Hashmi, in Silver in Organic Chemistry, ed. M. Harmata, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, ch. 12, pp. 357–379 Search PubMed.
- The first derivative of reaction of intermediate II, path B, after the reaction with nitromethane is most probably a transient alcholate anion. Altough alcholate anion species are known to add across triple bonds, all experimental data in our hands seems to point out that nitroalcohol 11, once formed, is not prone to undergo cycloisomerisation to afford isochromene 12 despite the weak basic conditions of our catalytic system.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- F. Bellina, D. Ciucci, P. Vergamini and R. Rossi, Tetrahedron, 2000, 56, 2533–2545 CrossRef CAS.
- L. Pérez, A. Pinazo, M. Teresa García, M. Lozano, A. Manresa, M. Angelet, M. Pilar Vinardell, M. Mitjans, R. Pons and M. Rosa Infante, Eur. J. Med. Chem., 2009, 44, 1884–1892 CrossRef PubMed.
- Q. Huang, J. A. Hunter and R. C. Larock, J. Org. Chem., 2002, 67, 3437–3444 CrossRef CAS PubMed.
- R. Bukšnaitienė, A. Urbanaitė and I. Čikotienė, J. Org. Chem., 2014, 79, 6532–6553 CrossRef PubMed.
- D. Yue, N. Della Cá and R. C. Larock, J. Org. Chem., 2006, 71, 3381–3388 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Yields and 1H NMR tabulated spectra for all known compounds. Figures reporting full NMR spectra for all compounds. See DOI: 10.1039/c6ra22231e |
| This journal is © The Royal Society of Chemistry 2016 |