
Selective ring opening of methylcyclopentane over surface-decorated Ir–Co bimetallic catalysts synthesized by galvanic replacement reaction
Zhen Wang†
,
Aiguo Zheng†,
Renyang Zheng*,
Mingfeng Li,
Huifeng Li,
Guangtong Xu and
Guofu Xia*
Research Institute of Petroleum Processing (RIPP), SINOPEC, 18 Xueyuan Road, Haidian District, Beijing 100083, China. E-mail: zhengrenyang.ripp@sinopec.com; xiaguofu.ripp@sinopec.com
First published on 24th October 2016
Abstract
Surface-decorated Ir–Co bimetallic catalysts were synthesized by a modified galvanic replacement reaction and evaluated by the selective ring opening (SRO) of methylcyclopentane (MCP). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was utilized to characterize the catalyst synthesis process, and H2-temperature-programmed reduction (H2-TPR) was performed to characterize the reduction behavior of the calcined catalysts and oxidation resistance of the reduced catalysts. Transmission electron microscopy (TEM) analysis was used to characterize metallic particle size distribution. The reactor evaluation results show quite different behaviors in conversion and product selectivity of the surface-decorated Ir–Co/SiO2 and monometallic Ir/SiO2. At optimized Ir content, MCP conversion and mass-specific rate of SRO are significantly higher when catalyzed by surface-decorated Ir–Co/SiO2 than by monometallic Ir/SiO2. Moreover, compared with Ir/SiO2, Ir–Co/SiO2 has decreased 2-methylpentane selectivity but improved n-hexane selectivity. These catalytic behaviors were found to correlate with the surface-decorated bimetallic structure consisting of Co/SiO2 decorated with surface Ir atoms.
1. Introduction
The selective ring-opening (SRO) process of naphthenic compounds, during which only an endocyclic C–C bond is cleaved, leaving the number of carbon atoms in the reaction products unchanged, has great importance for improving the cetane number of diesel fuel.1–4 Numerous studies have shown good catalytic performance for SRO by using the noble metal catalysts (e.g. Ir, Pt, Ru, Rh).5–9 There are three main mechanisms10,11 for the ring-opening process of naphthenes catalyzed by noble metals: the dicarbene mechanism, multiplet mechanism and metallocyclobutane mechanism. When SRO reaction occurs according to the dicarbene mechanism, naphthenic compounds adsorb to the catalyst active sites perpendicularly via two carbon atoms of the naphthenic ring, which preferentially induces the breakage of C–C bond at the unsubstituted position of the ring.12,13 However, if the C–C bond is cleaved at the substituted position of the ring by the multiplet mechanism or the metallocyclobutane mechanism, the cetane number can be improved.10,14Among the various noble metals, Ir is proven to be the most active and selective ring-opening catalyst, promoting the SRO reaction of naphthenes through the dicarbene mechanism.15–19 However, Ir is one of the rarest and most valuable elements on earth. Therefore, many attempts have been made to improve the catalytic performance of Ir and reduce the catalyst cost. González-Cortés et al.20 reported a modified Ir-containing catalyst with the addition of potassium to improve the ring-opening selectivity for 1,3-dimethylcyclohexane. Shen et al.21 synthesized bimetallic Pd–Ru catalyst for SRO of indane, which displayed the same high single-cleavage selectivity as Ir and was envisioned as a viable alternative to monometallic Ir.
Apart from the above methods, another desirable bimetallic catalyst structure, with active Ir atoms dispersed on the nanoparticle surfaces of the inexpensive metal (e.g. Co, Ni), will allow more Ir atoms to be exposed to the reactants and promote the SRO reaction. Ziaei-Azad et al.22 synthesized bimetallic core–shell structured nanoparticles with Ni atoms in the particle core and Ir atoms on the surface by hydrogen-sacrificial technique, and then loaded the nanoparticles onto the support for the SRO reaction of indane, significantly improving the catalytic activity compared to the monometallic Ir. As a model reaction, the SRO of MCP on supported metal catalysts has been extensively studied.23–25 Since ring opening of MCP on Ni catalyst is usually accompanied by extensive secondary cracking of the primary C6-alkane hydrogenolysis products to C1–C5 paraffins,1 in the current work, we selected Co nanoparticles to support the admetal Ir in order to avoid the excessive hydrogenolysis. Thus, the above leading works motivate us to explore surface-decorated Ir–Co bimetallic catalysts synthesized via a modified galvanic replacement,26,27 an improved method based on the traditional impregnation method. Several characterization methods, including inductively coupled plasma atomic emission spectroscopy (ICP-AES), H2-temperature-programmed reduction (H2-TPR) and transmission electron microscopy (TEM), were employed to verify the structures and compositions of the catalysts and understand their catalytic behavior in the SRO reaction of MCP.
2. Experimental methods
2.1. Catalyst preparation
The surface-decorated Ir–Co bimetallic catalysts were synthesized by a modified galvanic replacement in this study. The precursor Co(NO3)2·6H2O was dissolved in deionized water completely and then impregnated onto the SiO2 support. After the impregnation process, the product was dried at 120 °C for 4 h and then calcined in air at 300 °C for 3 h to produce the calcined Co/SiO2. The calcined Co/SiO2 was reduced by H2, cooled down to room temperature, and subsequently put into the aqueous solution of IrCl3·xH2O for continuous shaking. It should be pointed out that the reduced catalyst was put directly into IrCl3·xH2O solution, and the treatment of the reduced catalyst by flushing N2 to remove the adsorbed H2 has no obvious influence on the catalytic performance. Then, the supernatant was removed, and the resultant catalysts were washed and dried to obtain the surface-decorated bimetallic catalysts x%Ir–10%Co/SiO2, where x stands for the nominal weight ratio of Ir to the catalyst (x = 0.1, 0.3 and 1.0). The Co content of the bimetallic samples is 10 wt%, with the amount of metallic Co replaced by Ir being neglected.For comparison, a traditional co-impregnation (CI) method was used to synthesize 1%Ir–10%Co/SiO2-CI catalyst. The precursors of IrCl3·xH2O and Co(NO3)2·6H2O were dissolved in deionized water completely and impregnated onto the SiO2 support. After the impregnation process, the catalyst was dried at 120 °C for 4 h and then calcined in air at 300 °C for 3 h to produce calcined 1%Ir–10%Co/SiO2-CI. The corresponding monometallic Ir/SiO2 and Co/SiO2 catalysts were also synthesized by impregnation method to serve as control samples.
2.2. Catalyst characterization
2.3. Catalytic evaluation
Studies of SRO reaction for MCP were performed in a continuous flow, fixed-bed microreactor under 280 °C and 2.0 MPa. Before the reaction, 0.15 g of catalyst mixed with quartz powder was reduced by H2 at 350 °C (450 °C for monometallic Co/SiO2) for 1 h in the microreactor. Through the reduction process, the catalyst was completely reduced. During the reaction, the feedstock of MCP, with a liquid flow rate of 0.1 mL min−1, was carried to the reactor by a constant flow pump. The gas flow of H2 was 400 mL min−1. The products were analyzed by an online Agilent 7890 GC equipped with a flame ionization detector (FID).3. Results and discussion
3.1. Characterization of synthesis process by ICP-AES
During the synthesis of surface-decorated Ir–Co/SiO2, the different reduction potentials (1.16 V for the Ir3+/Ir pair and −0.28 V for the Co2+/Co pair) produce the driving force for the galvanic replacement reaction.28,29 When reduced Co/SiO2 was put into the Ir3+ solution, it was assumed that a galvanic replacement reaction occurred between Ir3+ and Co, leading to the preferential deposition of Ir on the surfaces of Co nanoparticles instead of SiO2 support (Scheme 1). To verify the above assumption, the concentration of Ir3+ before and after galvanic replacement during the synthesis procedure was measured (Table 1). Only trace amount of Ir3+ (<15 mg L−1) remains in the supernatants after the galvanic replacement reaction, and the calculated loading ratio of Ir3+ on the x%Ir–10%Co/SiO2 catalyst is more than 90%, implying that most of the Ir3+ in the aqueous precursor solution is loaded onto the catalyst. | ||
| Scheme 1 Illustration of the modified galvanic replacement from Co/SiO2 to Ir–Co/SiO2. | ||
| Items | 0.1%Ira | 0.3%Ira | 1%Ira |
|---|---|---|---|
| a For x%Ir (x = 0.1, 0.3 and 1), x stands for the nominal weight ratio of Ir to Ir–Co/SiO2.b Catalyst synthesis via the galvanic replacement reaction.c Comparative synthesis without the Co/SiO2 reduction procedure. | |||
| Ir3+ concentration in aqueous precursor solution (before galvanic replacement)/mg L−1 | 55.7 | 167 | 562 |
| Ir3+ concentration in the supernatants (after galvanic replacement)b/mg L−1 | <1 | 14.5 | 14.2 |
| Loading ratio of Ir3+ on the catalystb | 99.9% | 91.3% | 97.5% |
| Ir3+ concentration in the supernatants (comparative samples)c/mg L−1 | 32.0 | 154 | 569 |
Furthermore, a comparative experiment was performed to distinguish the distribution and state of the Ir species loaded onto the x%Ir–10%Co/SiO2 catalyst, whether in the form of Ir3+ via adsorption or metallic Ir via galvanic replacement reaction. The calcined 10% Co/SiO2, without the reduction process, was directly put into the aqueous precursor solution of IrCl3·xH2O with continuous shaking, then the supernatants were collected for ICP-AES analysis. The data in the second and last rows of Table 1 show little difference in Ir3+ concentration between the supernatants and aqueous precursor solution, indicating that only trace amount of Ir3+ is loaded onto the catalyst via adsorption. Therefore, it can be deduced that by the galvanic replacement reaction, most of the Ir3+ in the aqueous precursor solution is preferentially deposited onto the surfaces of Co nanoparticles in the form of metallic Ir instead of Ir3+ adsorbed by SiO2 support.
3.2. H2-TPR results
The reduction behavior of the calcined catalysts by TPR is shown in Fig. 1(a). The Co/SiO2 shows two main reduction peaks at about 285 °C and 320 °C, which attribute to the successive reduction of Co3O4 to CoO and CoO to metallic Co, respectively.30,31 As shown in Fig. 1(a), Ir can be reduced at about 163 °C, lower than that of Co. Thus for Ir–Co/SiO2-CI, hydrogen that dissociates on the Ir surface could migrate to the surface of Co3O4 and support, resulting in Co being reduced at much lower temperatures. These results also suggest that the catalysts were completely reduced by H2 at 350 °C (450 °C for monometallic Co/SiO2). | ||
| Fig. 1 TPR profiles of (a) the calcined catalysts and (b) the “reduced-oxidized” catalysts. The signal of 1%Ir/SiO2 in (a) has been magnified 5 times. The “reduced-oxidized” catalysts were analyzed by a modified TPR process: the catalysts were reduced, cooled down to room temperature, exposed to the air, and then used for the TPR experiment. | ||
To further verify the surface compositions of the catalysts, oxidation resistance experiments for the reduced catalysts were performed via a modified TPR process, the results of which are shown in Fig. 1(b). The H2-TPR analysis of “Ir/SiO2 (reduced-oxidized)” shows an almost-flat profile, indicating that reduced Ir is nearly not oxidized in the air atmosphere at room temperature. For “10%Co/SiO2 (reduced-oxidized),” the intense reduction peak around 180 °C can be attributed to the reduction of CoO, the oxidation state of Co at room temperature. Specially, for the three curves of “x%Ir–10%Co/SiO2 (reduced-oxidized)” (x = 0.1, 0.3 and 1.0), the intensity of reduction peaks for “CoO→Co” decreases compared to that for “10%Co/SiO2 (reduced-oxidized).” These indicate that for the surface-decorated Ir–Co/SiO2 catalysts, Ir atoms decorated on the surfaces of Co nanoparticles via galvanic replacement suppress the oxidation of Co at room temperature.
3.3. TEM results
High-angle annular dark field (HAADF) TEM images and particle size distributions are shown in Fig. 2. Particle size distributions of the supported bimetallic nanoparticles, assuming a spherical shape, were obtained from the measurement of about 300 particles found in several arbitrarily chosen areas of enlarged micrographs. The dominant particle size in each catalyst appears to range in diameter from approximately 2 nm to 5 nm. The average diameter is 3.4, 3.2 and 2.9 nm for 0.3%Ir–10%Co/SiO2, 1%Ir–10%Co/SiO2 and 1%Ir–10%Co/SiO2-CI, respectively.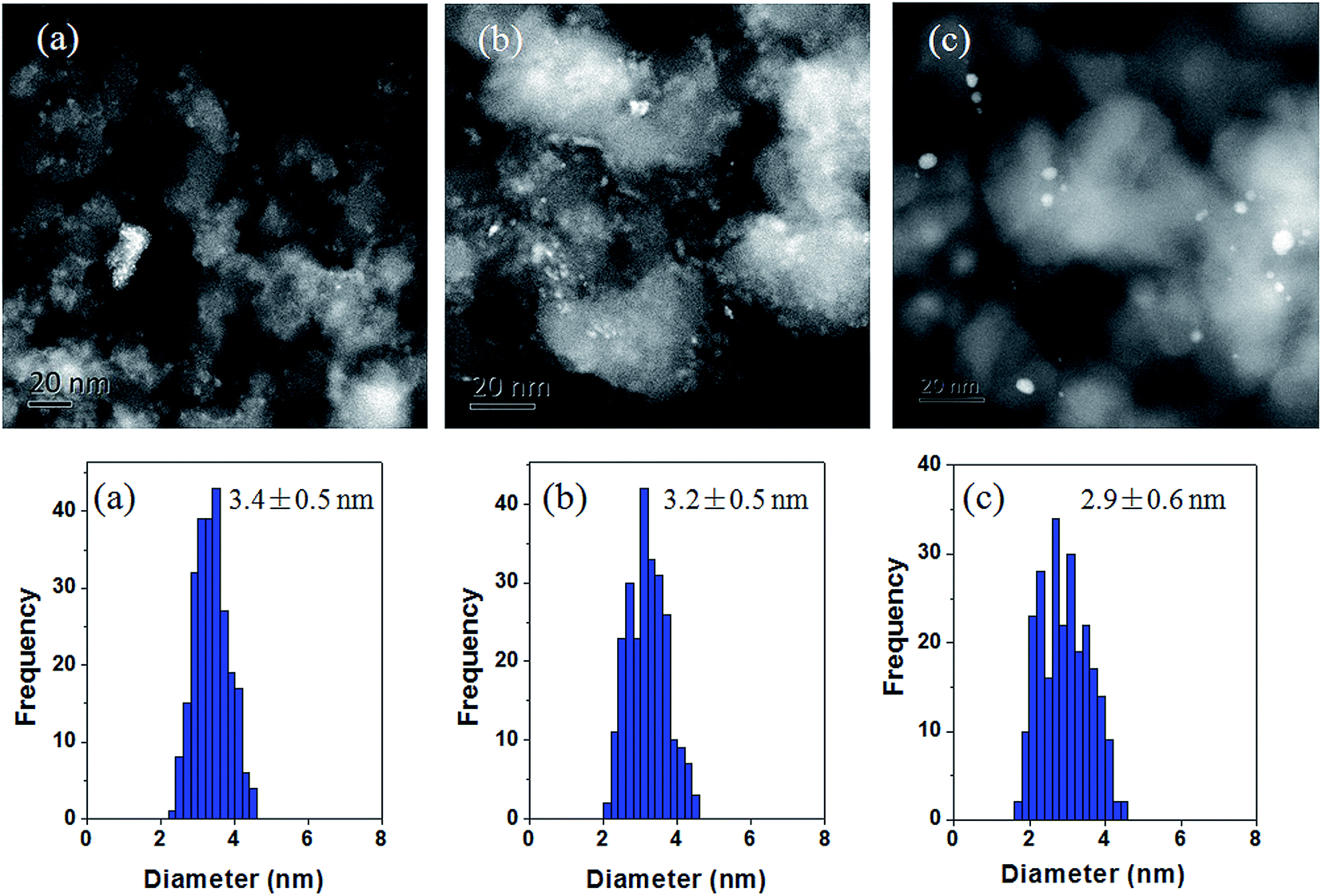 | ||
| Fig. 2 TEM images and particle size distributions for (a) 0.3%Ir–10%Co/SiO2, (b) 1%Ir–10%Co/SiO2 and (c) 1%Ir–10%Co/SiO2-CI. | ||
Furthermore, STEM-EDX was used to measure the distributions of Ir and Co in the 1%Ir–10%Co/SiO2 catalyst, as shown in Fig. 3. The images obtained by three scanning modes, including a selected point, selected line scan and selected area scan (Fig. 3(a–c)), show that all the bright nanoparticles are composed of Co atoms, while some brighter nanoparticles are composed of both Co and Ir atoms. Combining the STEM and HRTEM images (Fig. 3(d)), the distribution of Ir atoms is clearly identified as single atoms or clusters (<1 nm) situated at surfaces of Co nanoparticles.
 | ||
| Fig. 3 Representative STEM images in (a) a selected point, (b) selected line scan mode, (c) selected area scan mode (mapping), and (d) HRTEM images of 1%Ir–10%Co/SiO2 catalyst. The green signals stand for Co and blue signals for Ir in (b) and (c). | ||
3.4. Catalytic evaluation
Flow reactor studies of SRO reaction for MCP were employed to compare the catalytic performance of different catalysts (Table 2). No detectable conversion of MCP is observed over monometallic Co/SiO2. For the monometallic Ir/SiO2 catalysts, the increased Ir content from 0.1 wt% to 1 wt% gives a significant improvement of MCP conversion. On the other hand, for the surface-decorated x%Ir–10%Co/SiO2, with the increase of Ir content, the MCP conversion displays a volcano trend and reaches its maximum value at 0.3 wt%. It can be inferred that during the synthesis of x%Ir–10%Co/SiO2, the increased Ir3+ concentration in aqueous precursor solution accelerates the reaction rate of galvanic replacement, leading to the non-uniform deposition of Ir atoms on the Co nanoparticle surfaces. Thus, the non-uniform deposition of Ir atoms restrains the catalytic behavior of 1%Ir–10%Co/SiO2. Moreover, Fig. 4 displays the mass-specific rate for SRO of MCP as a function of Ir content over monometallic Ir/SiO2 and surface-decorated bimetallic Ir–Co/SiO2. Increasing Ir content improves the mass-specific rate for monometallic Ir/SiO2 but decreases it for Ir–Co/SiO2 bimetallic catalysts. These suggest that SRO is sensitive to Ir content and the surface structure of bimetallic catalysts.| Catalysts | Conversion (%) | RO selectivityb (%) | 2MP/3MP | |||
|---|---|---|---|---|---|---|
| SUM | 2MP | 3MP | n-H | |||
| a Reaction conditions: catalyst 0.15 g, pressure 2.0 MPa, temperature 280 °C.b 2MP: 2-methylpentane; 3MP: 3-methylpentane; n-H: n-hexane; SUM: the sum of 2MP, 3MP and n-H. | ||||||
| 0.1%Ir/SiO2 | 2.8 | 99.8 | 70.5 | 28.9 | 0.4 | 2.4 |
| 0.1%Ir–10%Co/SiO2 | 18.3 | 77.2 | 46.0 | 28.1 | 3.1 | 1.6 |
| 0.3%Ir/SiO2 | 10.5 | 98.6 | 70.3 | 28.1 | 0.2 | 2.5 |
| 0.3%Ir–10%Co/SiO2 | 21.9 | 76.9 | 46.8 | 27.1 | 3.0 | 1.7 |
| 1%Ir/SiO2 | 75.0 | 99.1 | 71.7 | 27.1 | 0.3 | 2.6 |
| 1%Ir–10%Co/SiO2 | 12.7 | 79.8 | 47.8 | 28.8 | 3.2 | 1.7 |
| 1%Ir–10%Co/SiO2-CI | 0.4 | 100 | 69.0 | 29.5 | 1.5 | 2.3 |
| 10%Co/SiO2 | 0 | — | — | — | — | — |
 | ||
| Fig. 4 Mass-specific rate as a function of Ir content for monometallic Ir/SiO2 and surface decorated bimetallic Ir–Co/SiO2. | ||
For 0.1 wt% and 0.3 wt% Ir content, MCP conversion (Table 2) and mass-specific rate of SRO (Fig. 4) are much higher when catalyzed by surface-decorated Ir–Co/SiO2 than by monometallic Ir/SiO2. It could be explained that for Ir–Co/SiO2, the catalytically active Ir atoms are highly dispersed on the Co nanoparticle surfaces via the galvanic replacement reaction, thus significantly improving the dispersion and utilization of Ir compared to Ir/SiO2. A similar trend has also been observed on the SRO of indane over Ir–Ni bimetallic catalysts reported by Ziaei-Azad et al.22 With the increase of Ir content to 1 wt%, the 1%Ir–10%Co/SiO2 catalyst does not have an advantage compared to 1%Ir/SiO2. At the same time, compared with another two catalysts with 1 wt% Ir (1%Ir–10%Co/SiO2 and 1%Ir/SiO2), 1%Ir–10%Co/SiO2-CI shows the least catalytic activity for SRO of MCP. The content of Co in 1%Ir–10%Co/SiO2-CI is much higher than that of Ir, with the Co/Ir atomic ratio of 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Therefore, one possible reason is that most Ir atoms in 1%Ir–10%Co/SiO2-CI are covered by Co atoms through the co-impregnation synthesis procedure and not exposed on the catalyst surfaces.
:1. Therefore, one possible reason is that most Ir atoms in 1%Ir–10%Co/SiO2-CI are covered by Co atoms through the co-impregnation synthesis procedure and not exposed on the catalyst surfaces.
The SRO reaction of MCP produces n-hexane (n-H), 2-methylpentane (2MP) and 3-methylpentane (3MP) (Scheme 2). Previous research studies have demonstrated that SRO reaction occurs according to a multiplet mechanism, a dicarbene mechanism and a metallocyclobutane mechanism over different noble metals.1,10,11,32 The multiplet mechanism is usually observed over Pt catalysts, with an equal probability of breaking endocyclic bonds, producing the statistical ratio of 2MP:3MP:n-H = 2:1:2.10,33 The dicarbene mechanism often happens over Ir catalysts, which preferentially induces endocyclic C–C bond breakage at the unsubstituted position of the ring, leading to the statistical ratio of 2MP:3MP = 2:1 and trace amounts of n-H.12,13 In addition to these two typical mechanisms for SRO reaction, Gault et al.34,35 proposed an alternative one that would operate through a metallocyclobutane intermediate consisting of a metal atom and three C atoms, which would allow the breakage of substituted C–C bonds if an external methyl group were involved in the intermediate.
 | ||
| Scheme 2 Ring-opening products of MCP. | ||
The ring-opening (RO) selectivity, product distribution and molar ratio of 2MP/3MP are listed in Table 2. The results of Ir/SiO2 in the present work are consistent with previous research work showing that the hydrogenolysis of MCP over Ir-based catalysts is selective to the formation of 2MP and 3MP.1,17 More interestingly, the surface-decorated Ir–Co/SiO2 and monometallic Ir/SiO2 show quite different behaviors in terms of product selectivity. The RO selectivity over Ir–Co/SiO2 is about 76–80%, lower than the 99% over Ir/SiO2. The cracking products on Ir–Co/SiO2 mainly include methane, butane and 2-methyl-butane, as analysed by gas chromatography-mass spectrometry (GC-MS). Meanwhile, compared with Ir/SiO2, Ir–Co/SiO2 shows a lower selectivity toward 2MP (∼47% vs. 71%), corresponding to lower 2MP/3MP ratio (∼1.6 vs. 2.5) but higher selectivity toward n-H (∼3.1% vs. 0.3%). Therefore, it can be inferred that the SRO reaction of MCP over Ir–Co/SiO2 follows a different mechanism compared to monometallic Ir/SiO2.
As reported by Chen and other research groups,36–40 bimetallic catalysts often show electronic and chemical properties that differ distinctly from those of the parent metals. For example, it has been demonstrated that Pt-terminated Pt–Co–Pt(111) surface, which represents a subsurface bimetallic structure with Pt on the top-most surface layer and Co residing in the subsurface region, shows much higher activity for the hydrogenation of cyclohexene than Co-terminated Co–Pt–Pt(111) and the corresponding monometallic surfaces.41 The catalytic hydrogenation pathway on the subsurface bimetallic structures has been correlated to the modification of the electronic properties of Pt by the subsurface Co atoms.42 It is possible that such effect also plays a role in the SRO reaction of MCP over the surface-decorated Ir–Co/SiO2 bimetallic catalysts. Moreover, X-ray photoelectron spectroscopy (XPS) should shed light on the electronic effects of surface-decorated structure. However, at the lower Ir content, the overlapping of Ir 4f and Co 3p levels makes it very difficult to determine the exact binding energy shift.43 More detailed density functional theory (DFT) calculations would be needed to further understand the electronic properties.
4. Conclusions
We explored the utilization of surface-decorated Ir–Co bimetallic catalysts synthesized via galvanic replacement for the selective ring-opening of MCP. Results from ICP-AES show that by the galvanic replacement reaction, most of the Ir3+ in the precursor solution is preferentially deposited onto the surfaces of Co nanoparticles in the form of metallic Ir instead of Ir3+ adsorbed by SiO2 support. The modified TPR results show that Ir atoms decorated on the surfaces of Co nanoparticles via galvanic replacement suppress the oxidation of Co at room temperature. TEM measurements provide additional information on the metallic particle size distribution, and the combination of STEM-EDX and HRTEM images identifies the single atoms or clusters of Ir situated at the surface of Co nanoparticles. At optimized Ir content, MCP conversion and mass-specific rate of SRO are significantly higher when catalyzed by surface-decorated Ir–Co/SiO2 than by monometallic Ir/SiO2. Moreover, compared with Ir/SiO2, Ir–Co/SiO2 decreases 2-methylpentane selectivity but improves the n-hexane selectivity, which is attributed to the surface-decorated bimetallic structure consisting of Co/SiO2 decorated with surface Ir atoms. Results from the current study also identify research opportunities in synthesizing supported bimetallic catalysts by surface decoration via galvanic replacement.Acknowledgements
This work was supported by the Major State Basic Research Development Program (Grant No. 2012CB224802) and SINOPEC Research Project (Grant No. 115059).References
- G. B. McVicker, M. Daage, M. S. Touvelle, C. W. Hudson, D. P. Klein, W. C. Baird, B. R. Cook, J. G. Chen, S. Hantzer and D. Vaughan, J. Catal., 2002, 210, 137–148 CrossRef CAS.
- L. Di Felice, N. Catherin, L. Piccolo, D. Laurenti, E. Blanco, E. Leclerc, C. Geantet and V. Calemma, Appl. Catal., A, 2016, 512, 43–51 CrossRef CAS.
- D. W. Flaherty, A. Uzun and E. Iglesia, J. Phys. Chem. C, 2015, 119, 2597–2613 CAS.
- D. W. Flaherty, D. D. Hibbitts and E. Iglesia, J. Am. Chem. Soc., 2014, 136, 9664–9676 CrossRef CAS PubMed.
- A. Haas, S. Rabl, M. Ferrari, V. Calemma and J. Weitkamp, Appl. Catal., A, 2012, 425, 97–109 CrossRef.
- D. Teschner, D. Duprez and Z. Paál, J. Mol. Catal. A: Chem., 2002, 179, 201–212 CrossRef CAS.
- A. Djeddi, I. Fechete and F. Garin, Appl. Catal., A, 2012, 413, 340–349 CrossRef.
- S. A. D'Ippolito, C. Especel, F. Epron and C. L. Pieck, Fuel Process. Technol., 2015, 140, 180–187 CrossRef.
- H. Shi, O. Y. Gutiérrez, A. Zheng, G. L. Haller and J. A. Lercher, J. Phys. Chem. C, 2014, 118, 20948–20958 CAS.
- H. Du, C. Fairbridge, H. Yang and Z. Ring, Appl. Catal., A, 2005, 294, 1–21 CrossRef CAS.
- P. T. Do, W. E. Alvarez and D. E. Resasco, J. Catal., 2006, 238, 477–488 CrossRef CAS.
- F. G. Gault, Adv. Catal., 1981, 30, 1–95 CAS.
- R. Moraes, K. Thomas, S. Thomas, S. Van Donk, G. Grasso, J. Gilson and M. Houalla, J. Catal., 2012, 286, 62–77 CrossRef CAS.
- Z. Zhao, L. V. Moskaleva and N. Rösch, J. Catal., 2013, 299, 146–149 CrossRef CAS.
- A. Piegsa, W. Korth, F. Demir and A. Jess, Catal. Lett., 2012, 142, 531–540 CrossRef CAS.
- S. Lecarpentier, J. van Gestel, K. Thomas, J. Gilson and M. Houalla, J. Catal., 2008, 254, 49–63 CrossRef CAS.
- P. Samoila, M. Boutzeloit, C. Especel, F. Epron and P. Marecot, Appl. Catal., A, 2009, 369, 104–112 CrossRef CAS.
- F. Locatelli, J. Candy, B. Didillon, G. P. Niccolai, D. Uzio and J. Basset, J. Am. Chem. Soc., 2001, 123, 1658–1663 CrossRef CAS PubMed.
- C. J. Hagedorn, M. J. Weiss, T. W. Kim and W. H. Weinberg, J. Am. Chem. Soc., 2001, 123, 929–940 CrossRef CAS PubMed.
- S. L. González-Cortés, S. Dorkjampa, P. T. Do, Z. Li, J. M. Ramallo-López and F. G. Requejo, Chem. Eng. J., 2008, 139, 147–156 CrossRef.
- J. Shen and N. Semagina, ACS Catal., 2013, 4, 268–279 CrossRef.
- H. Ziaei-Azad and N. Semagina, ChemCatChem, 2014, 6, 885–894 CrossRef CAS.
- P. Samoila, M. Boutzeloit, C. Especel, F. Epron and P. Marécot, J. Catal., 2010, 276, 237–248 CrossRef CAS.
- D. Teschner, K. Matusek and Z. Paál, J. Catal., 2000, 192, 335–343 CrossRef CAS.
- D. P. Upare and C. W. Lee, Fuel Process. Technol., 2014, 126, 243–248 CrossRef CAS.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- S. L. Yang, C. Y. Cao, F. F. Wei, P. P. Huang, Y. B. Sun and W. G. Song, ChemCatChem, 2014, 6, 1868–1872 CrossRef CAS.
- S. Wang, J. A. He, J. Xie, Y. Zhu, Y. Xie and J. G. Chen, Appl. Surf. Sci., 2008, 254, 2102–2109 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, New York, 15th edn, 1999 Search PubMed.
- E. van Steen, G. S. Sewell, R. A. Makhothe, C. Micklethwaite, H. Manstein, M. de Lange and C. T. O'Connor, J. Catal., 1996, 162, 220–229 CrossRef CAS.
- A. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486–499 CrossRef.
- Z. Zhao, L. V. Moskaleva and N. Rösch, ACS Catal., 2013, 3, 196–205 CrossRef CAS.
- G. Maire, G. Plouidy, J. C. Prudhomme and F. G. Gault, J. Catal., 1965, 4, 556–569 CrossRef CAS.
- V. Amirebrahimi, A. Choplin, P. Parayre and F. G. Gault, Nouv. J. Chim., 1980, 4, 431–435 CAS.
- P. T. Do, W. E. Alvarez and D. E. Resasco, J. Catal., 2006, 238, 477–488 CrossRef CAS.
- R. Zheng, M. D. Porosoff, J. L. Weiner, S. Lu, Y. Zhu and J. G. Chen, Appl. Catal., A, 2012, 419, 126–132 CrossRef.
- J. H. Sinfelt, Acc. Chem. Res., 1977, 10, 15–20 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281–294 CrossRef CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- R. Zheng, M. P. Humbert and Y. Z. J. Chen, Catal.: Sci. Technol., 2011, 1, 638–643 CAS.
- M. P. Humbert and J. G. Chen, J. Catal., 2008, 257, 297–306 CrossRef CAS.
- N. A. Khan, L. E. Murillo and J. G. Chen, J. Phys. Chem. B, 2004, 108, 15748–15754 CrossRef CAS.
- W. Cai, N. Homs and P. R. de la Piscina, Green Chem., 2012, 14, 1035–1043 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |