
One-step hydrothermal synthesis of novel Ag3VO4/Ag4V2O7 composites for enhancing visible-light photocatalytic performance
Abstract
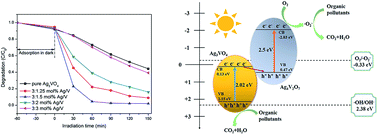
Novel direct Z-scheme Ag3VO4/Ag4V2O7 composites were synthesized by one-step hydrothermal method for the first time. The photocatalytic performance was evaluated by the degradation of methylene blue (MB) and phenol under visible light irradiation (λ ≥ 420 nm). The results showed that the as-synthesized heterojunctions can significantly enhance photocatalytic activity in comparison with pure Ag3VO4. The optimum photocatalytic efficiency of 3 : 1.5 mol% Ag/V sample for the degradation MB and phenol reached up to about 100% and 62.1%, respectively. In addition, 3 : 1.25 mol% Ag/V sample had an excellent visible light response range, which occurred around 652 nm. Based on the photocurrent and the radical-trapping experiments, the possible for the enhancing photocatalytic mechanism was found to be a direct Z-scheme heterojunction system, which not only can improve the photogenerated electron–hole pairs' separation but also exhibit strong oxidation and reduction ability.
 Please wait while we load your content...
Please wait while we load your content...

