
Synthesis of a BiFeO3 nanowire-reduced graphene oxide based magnetically separable nanocatalyst and its versatile catalytic activity towards multiple organic reactions†
Debabrata Moitra,
Barun Kumar Ghosh,
Madhurya Chandel and
Narendra Nath Ghosh*
Nanomaterials Lab, Department of Chemistry, Birla Institute of Technology and Science, Pilani K. K. Birla Goa Campus, Zuarinagar, Goa 403726, India. E-mail: naren70@yahoo.com; Fax: +91 832 2557033; Tel: +91 832 2580318
First published on 10th October 2016
Abstract
Herein, we report for the first time synthesis of a BiFeO3 nanowire-reduced graphene oxide nanocatalyst (BFO–RGO) using a hydrothermal method. The BFO–RGO nanocatalyst exhibited excellent catalytic activity towards Biginelli reaction, Click reaction, styrene epoxidation, 4-NP reduction and a herbicide, (trifluralin) reduction. The novelty of this catalyst lies in its high catalytic efficiency towards many organic reactions, easy separation and good reusability.
1. Introduction
Development of a novel catalyst, which can exhibit high catalytic activity towards various organic synthesis reactions, is strategically very important for applications in various pharmaceutical and chemical industries. In this paper, we report the synthesis of a novel but versatile catalyst for the following organic reactions: (i) Biginelli reaction, (ii) synthesis of 1,2,3-triazole derivatives by Click reaction, (iii) epoxidation of styrene and (iv) reduction of 4-nitrophenol (4-NP) and a herbicide, (trifluralin).Importance of Biginelli reaction lies in its wide applications for preparation of various drugs for cardiovascular diseases (e.g. nifedipine), antihypertensivity, potent HIV-group-120 CD4 inhibitor etc. in pharmaceutical industries.1–4 Conventional Biginelli reactions suffer from low yield of products, requirement of high temperature, prolonged reaction time, usage of strong Lewis acid catalysts, their separation etc.1,2,5–7 Synthesis of 1,2,3-triazoles via ‘Click reaction’ has gained considerable attention because of its applications in preparation of pharmaceutical agents, agrochemicals, dyes photostabilizers etc. However, sometimes lack of sensitivity, low yields and requirement of elevated reaction temperature are also associated limitations of this reaction.8–10 Synthesis of styrene oxide, which is one of the commercially important resins, via epoxidation of vinyl group of styrene is a common technique. However, use of peracid as catalyst is a major drawback of this process, because peracids are not only corrosive and environmentally hazardous but also large amount of waste and by products generate in the reaction.11–18 Reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) is a very important reaction because nitrophenols and their derivatives are formed during the production of pesticides, herbicides, insecticides, synthetic dyes etc. and presence of 4-NP in the waste water discharged from industries causes water pollution. Moreover, 4-aminiphenol is an active precursor for various drugs (e.g. paracetamol, phenoceline etc.).19–21 Though several nanocatalysts, particularly metal nanoparticles (e.g. Au, Ag, Ru, Pd etc.),20–25 have been reported for reduction of 4-NP, but separation of the nanocatalysts is a major challenge.
Generally, nanoparticles exhibit high catalytic activity due to their large surface area.22 However, isolation and recovery of nanocatalyst demand special filtration methods or ultracentrifugation, which are tedious and inconvenient. Therefore, this difficulty of nanocatalyst recovery hampers their large scale usage, sustainability and economic viability. Moreover, there is always a chance of getting final product contaminated with unrecovered nanocatalyst. To overcome these issues, use of magnetic nanoparticle or introduction of magnetic character in the catalyst system appears as attractive solution. Magnetic nanocatalysts can simply and efficiently be removed from reaction mixtures by using an external magnetic field.
Several researchers have utilized Bi(III) salts as a homogeneous catalyst for various organic reactions such as Biginelli reaction, Click reaction etc.2,26,27 The presence of vacant d-orbital of Bi(III) helps to stabilize the intermediates in organic reactions.2 This factor makes Bi(III) an attractive catalyst. To the best of our knowledge, till date the catalytic properties of Bi(NO3)3, BiCl3 etc. has been exploited in various homogenous solution phase organic reactions.2,5,27 However, some inherent problems of homogenous catalyst, (such as catalyst separation, its reusability etc.), are also associated with Bi(III) catalyzed reactions. To overcome the above limitations, we have designed a BiFeO3 nanowire-reduced graphene oxide (RGO) containing heterogeneous catalyst which is expected to exhibit excellent catalytic activity towards all the above mentioned organic reactions and can also be magnetically separable from reaction mixture.
Several synthetic methods such as hydrothermal, co-precipitation, precursor based methods, microemulsion technique, sol–gel process, molten-salt method, conventional solid state method etc. have been reported for bismuth ferrite.28–33 However, till date synthesis of nanosized single-phase BiFeO3 is a challenging issue, because of volatility of Bi component at high temperature and formation of insoluble bismuth subnitrate or bismuthyl nitrate (Bi2O3·N2O5·2H2O)34,35 by hydrolysis of bismuth nitrate in aqueous medium. These factors seriously influence the formation of single-phase BiFeO3 and easily forming non-stoichiometric components (e.g. BiFe4O9, Bi12Fe0.63O18.945 etc.)30,33,34 are found to be present as impurity with BiFeO3. Though anodized aluminium oxide (AAO) template method, high voltage electrospinning method, surfactant and polymer auxiliary hydrothermal technique etc.36–39 have been reported for synthesis of BiFeO3 nanoparticle, but these methods are complicated in nature. To achieve enhanced magnetoelectric coupling and high electron mobility through ballistic charge transport mechanism Li et al. and Liu et al. have reported synthesis of BiFeO3 nanowires employing hydrothermal technique.34,40
Here, we report the synthesis of BiFeO3 nanowire-reduced graphene oxide (BFO–RGO) nanocatalysts, where BiFeO3 nanowires (BFO) are anchored on the surface of nanometer thin Reduced Graphene Oxide (RGO) sheets. In this catalyst, presence of graphene offers the following advantages: (i) high surface area for good dispersion of BFO which helps the reactant molecules to come in contact with catalytically active sites, (ii) two dimensional structures for adsorption of reactants, (iii) excellent electrical conductivity for electron transfers.13,14,19,25,41–45 Moreover, the hydroxyl, carbonyl or epoxy groups of RGO help to anchor BFO on its surface. On the other hand, BFO being a multiferroic material (ferroelectric and weakly ferromagnetic at room temperature) is capable of providing necessary magnetic character as well as catalytically active sites (due to presence of Bi3+ in BFO) in the BFO–RGO nanocatalyst. BFO nanowires are deemed to be more catalytically active than polycrystalline BFO,40,46 because of higher surface area of nanowires and more efficient ballistic charge transport along the wire axis than the diffusive transport in polycrystalline BFO nanoparticles.40 To the best of our knowledge, the synthesis of BiFeO3 nanowire-RGO nanocomposites and its application as a versatile catalyst for various organic reactions is not yet reported.
2. Experimental
2.1. Materials
Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O), iron(III) chloride hexahydrate (FeCl3·6H2O), acetone, ammonium hydroxide and 4-nitrophenol (4-NP) were purchased from Fischer Scientific, sodium hydroxide, sodium nitrate, sulphuric acid, potassium permanganate, urea and 30% H2O2 solution were purchased from Merck, India and benzaldehyde, phenylacetylene, ehyl acetoacetate, acetylacetone, styrene, styrene oxide, cyclohexene oxide, tert-butyl hydroperoxide (TBHP in 5–6 M decane), sodium borohydride (NaBH4), acetonitrile, trifluralin and graphite powder (mean particle size of <20 μm) were purchased from Sigma Aldrich and used without further purification. Distilled water was used throughout the experiment.2.2. Synthesis of graphene oxide (GO)
Graphene oxide was synthesized from graphite powder according to method reported by Hummers and Offeman.19,42,47 In this synthesis process, at first 1 g graphite and 0.6 g of NaNO3 were mixed with 35 ml of H2SO4 (18 M) at 0 °C. The mixture was stirred for 6 h. Then 3.8 g of KMnO4 was added to the suspension very slowly. The temperature of the solution rises to 35 °C and was maintained for 8 h so that complete oxidation takes place. Then 60 ml of distilled H2O was slowly added and the reaction temperature was increased to 98 °C. This temperature was maintained for 1 h and finally 2 ml of 30% H2O2 solution was added into the mixture and stirred for 0.5 h. The mixture was centrifuged and washed with 10% HCl solution and distilled H2O. The yellowish brown precipitate of graphene oxide was obtained and dried at 60 °C.2.3. Synthesis of BiFeO3-reduced graphene oxide nanocatalyst
We have employed a hydrothermal method to prepare BFO–RGO nanocatalyst. Synthesis of BFO–RGO were performed using the following steps: first step: 2.425 g Bi(NO3)3·5H2O and 1.352 g FeCl3·6H2O were mixed in 50 ml of acetone (99.8%) and sonicated for 30 min. Second step: an aqueous dispersion of GO (0.03189 g in 50 ml) was then added in the mixture. Third step: 150 ml of H2O and concentrated ammonia were added under vigorous stirring until the pH value of the solution reached 10–11. After filtering and rinsing with water, the red co-precipitate was redispersed in 40 ml water. Fourth step: under vigorous stirring, 5(M) NaOH aqueous solution was added into the suspension. Fifth step: this solution was transferred in a stainless steel autoclave with a Teflon liner and heated at 140 °C for 24 hours. The final black powder was separated from the reaction mixture and washed with distilled water followed by drying at 60 °C. We have also prepared pure BFO using same method without adding GO.2.4. Synthesis of 3,4-dihydropyrimidinone catalyzed by BFO–RGO nanocomposite
We have performed the synthesis of 3,4-dihydropyrimidinone (5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-one and 5-acetyl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one) in solventless condition as representative of Biginelli reaction. In typical synthesis, 1 mmol benzaldehyde (0.101 ml), 1.2 mmol urea (72 mg) and 1 mmol ethyl acetoacetate (0.126 ml) (or acetyl acetone (0.103 ml)) were mixed in a round bottom flask. To this mixture 25 mg of BFO–RGO catalyst was added and mixed thoroughly. The reaction mixture was then heated at 80 °C for 30 min. After cooling the reaction mixture was poured into ice cooled water and catalyst was separated by applying an external magnet. The product was then recrystallized. The same reactions were also conducted in presence of pure BFO and pure RGO.2.5. BFO–RGO catalyzed synthesis of 1,4-disubstituted 1,2,3-triazoles by ‘Click reaction’
For synthesis of 1,4-disubstituted 1,2,3-triazoles by ‘Click reaction’ BFO–RGO was used as catalyst. Here, 2-phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethanol and 2-(4-phenyl-[1,2,3]triazole-1-yl)-cyclohexanol were synthesized in aqueous medium as model reaction. In a typical synthesis, in a round bottomed flask 1 mmol styrene oxide (0.115 ml) (or, cyclohexene oxide (0.101 ml)), 1.1 mmol sodium azide (72 mg), 1 mmol phenyl acetylene (0.110 ml) and 25 mg of BFO–RGO were mixed with 3 ml water. This mixture was then refluxed at 80 °C for 2 h with constant stirring. Then the reaction mixture was allowed to cool down to room temperature and the catalyst was separated magnetically. The product was separated by filtration followed by recrystallization. The same reactions were also conducted in presence of pure BFO and pure RGO.2.6. Epoxidation of styrene catalyzed by BFO–RGO
The catalytic reaction for styrene epoxidation was carried in a 50 ml two-necked flask fitted with a reflux condenser. 50 mg of the BFO–RGO catalyst, 4 ml of acetonitrile and 5 mmol of styrene were added into the flask and stirred for 30 min under nitrogen atmosphere. Then 12.5 mmol of TBHP was added slowly under vigorous stirring and then the reaction mixture was heated at 100 °C temperature. Samples were periodically collected from the reaction mixture and analyzed by a gas chromatograph (GC-2014 Shimadzu) equipped with a capillary column (30 M × 0.25 mm × 0.25 mm) and a FID detector. The same reactions were also conducted in presence of pure BFO and pure RGO. It was observed that BFO–RGO acted as a catalyst with ∼79% conversion of styrene and ∼90% selectivity of styrene oxide after 5 h of reaction.2.7. Reduction of 4-NP and trifluralin catalyzed by BFO–RGO
To study the catalytic activity of BFO–RGO, reduction reactions were performed for 4-nitrophenol (4-NP) and trifluralin. In a typical run, 4.5 ml of 9 × 10−5 M aqueous solution of 4-NP (in case of trifluralin the concentration was 0.75 mM in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol and water mixture) was mixed with 0.5 ml·H2O and 1 ml 0.2 M NaBH4 solution. To this solution 2 ml aqueous suspension of the catalyst (0.4 g l−1) was added. 4 ml of this reaction mixture was immediately transferred in a quartz cuvette. The colored solution faded gradually as the reaction proceeded. The reaction was monitored by recording absorption spectrum by a UV-vis spectrophotometer (V-570, Jasco) at an interval of 1 min. UV-vis spectra of aqueous solution of 4-NP showed the maximum absorption peak (λmax) at 317 nm. This peak was red shifted to 400 nm after addition of NaBH4 due to formation of bright yellow colored 4-nitrophenolate ions. After addition of the catalyst gradually decrease of intensity of the peak at 400 nm indicated the progress of the reduction of 4-NP. Also, gradual development of small shoulder peak at 300 nm which is attributed to the absorption peak of 4-aminophenol (4-AP) was observed. For trifluralin λmax was 437 nm. The reaction was carried out at room temperature (30 ± 1 °C). All catalysis reactions were performed in triplicate. It is well documented that, metal oxide nanoparticle catalyzed reduction reaction of 4-NP in presence of excess NaBH4 proceed via pseudo first order kinetics.19,21,48,49 As the absorbance of 4-NP (or trifluralin) is proportional to its concentration, the ratio of absorbance of the 4-NP (or trifluralin) At (measured at time t) to Ao (at t = 0) is equal to Ct/Co (where Ct is the concentration of 4-NP (or trifluralin) at time t and Co is the initial concentration of the 4-NP (or trifluralin)). The apparent rate constant kapp was determined from the following equation:
:1 ethanol and water mixture) was mixed with 0.5 ml·H2O and 1 ml 0.2 M NaBH4 solution. To this solution 2 ml aqueous suspension of the catalyst (0.4 g l−1) was added. 4 ml of this reaction mixture was immediately transferred in a quartz cuvette. The colored solution faded gradually as the reaction proceeded. The reaction was monitored by recording absorption spectrum by a UV-vis spectrophotometer (V-570, Jasco) at an interval of 1 min. UV-vis spectra of aqueous solution of 4-NP showed the maximum absorption peak (λmax) at 317 nm. This peak was red shifted to 400 nm after addition of NaBH4 due to formation of bright yellow colored 4-nitrophenolate ions. After addition of the catalyst gradually decrease of intensity of the peak at 400 nm indicated the progress of the reduction of 4-NP. Also, gradual development of small shoulder peak at 300 nm which is attributed to the absorption peak of 4-aminophenol (4-AP) was observed. For trifluralin λmax was 437 nm. The reaction was carried out at room temperature (30 ± 1 °C). All catalysis reactions were performed in triplicate. It is well documented that, metal oxide nanoparticle catalyzed reduction reaction of 4-NP in presence of excess NaBH4 proceed via pseudo first order kinetics.19,21,48,49 As the absorbance of 4-NP (or trifluralin) is proportional to its concentration, the ratio of absorbance of the 4-NP (or trifluralin) At (measured at time t) to Ao (at t = 0) is equal to Ct/Co (where Ct is the concentration of 4-NP (or trifluralin) at time t and Co is the initial concentration of the 4-NP (or trifluralin)). The apparent rate constant kapp was determined from the following equation:| dCt/dt = −kappCt | (1) |
| ln(Ct/Co) = ln(At/Ao) = −kappt. | (2) |
The value of kapp was calculated from ln(At/Ao) vs. time plot.
After each cycle the catalyst was separated from the reaction mixture by applying external magnet. Then the catalyst was washed several times by deionized water and alcohol and dried for the next run. It was observed that no unreacted 4-NP (or trifluralin) molecule was remained adsorbed in the catalyst.
2.8. Characterization
Room temperature powder X-ray diffraction (XRD) pattern of the synthesized nanopowder was recorded using a powder X-ray diffractometer (Mini Flex II, Rigaku) with Cu Kα (λ = 0.15405 nm) radiation at a scanning speed of 2° min−1. High Resolution Transmission Electron Microscopy (HTEM) and SAED images of samples were obtained using JEOL JEM 1400. Fourier transform infrared spectra (FT-IR) were recorded in KBr by using FT-IR spectrophotometer (Shimadzu DR-8031). Raman spectra were taken on a Renishawin Via Raman microscope with a 633 nm laser excitation. Thermogravimetric analysis (TGA) analysis and differential scanning calorimetric were carried out using DTA-60 and DSC-60 (Shimaduzu). FESEM images of samples were obtained using Quanta 250 FEG (FEI). Energy dispersive X-ray spectra of the synthesized material were recorded using EDAX ELEMENT electron microscope. 1H NMR (nuclear magnetic resonance) spectra of the samples were recorded on a BRUKER 400 ULTRA SHIELD (400 MHz) instrument using deuterated solvent. Gas chromatograph was carried out using (Shimadzu GC-2014) equipped with a capillary column (30 M × 0.25 mm × 0.25 mm) and a FID detector. Room temperature magnetization with respect to external field was measured by using Vibrating Sample Magnetometer (VSM) (EV5, ADE technology). Multiple point BET surface area of the synthesized materials were measured with a surface area and porosity analyzer (Micromeritics Tristar 3000). Before the analysis, samples were degassed at 120 °C for 6 h. The purity of the products obtained by Biginelli reaction and Click reaction was verified using 1H NMR, FT-IR and melting point determination by DSC. (Details of spectral data of the synthesized compounds obtained from Biginelli, Click reaction and styrene epoxidation are provided in ESI (Fig. S1–S7†)).3. Results and discussion
3.1. Structure and morphology of BFO–RGO nanocomposites
Room temperature wide angle powder XRD was employed to identify the phases which were present in (i) the precipitate, formed due to the reaction of NH4OH with Bi(NO3)3 and FeCl3, (ii) the final product formed after hydrothermal treatment of precipitate in aqueous NaOH medium, (iii) the product when hydrothermal process was carried out in presence of the precipitate and GO in aqueous NaOH medium. XRD patterns are presented in Fig. 1. It was observed that, the reaction between NH4OH with Bi(NO3)3 and FeCl3 resulted in the formation of precipitate containing small particles of Fe(OH)3 and (Bi2O2)(OH)Cl (Fig. 1b). Pure BiFeO3 was formed due to hydrothermal treatment of this precipitate. The diffraction peaks of pure BFO (Fig. 1d) can be assigned to pure phase of BiFeO3 (JCPDS no. 20-0169), which indicates its rhombohedrally distorted perovskite structure with space group of R3c and lattice parameters of a = b = c = 5.62043 Å and α = β = γ = 59.35381°. These values are consistent with literature.34,40 XRD patterns of GO showed the diffraction peaks corresponding to (001) and (101) planes of GO13,19 (Fig. 1a). In case of BFO–RGO, diffraction peaks corresponding to pure BiFeO3 were observed (Fig. 1c). Here, absence of peaks for GO indicated that during preparation of BFO–RGO, GO flakes were converted to RGO and RGO sheets were exfoliated.19 The important point here is that, no impurity phase was detected. The synthetic method reported here showed its capability to produce BFO–RGO, where pure single phase BFO is present. According to the previously reported literature, synthesis of pure BiFeO3 is a challenge.43 Most of the reported methodologies produce either mixed phase BiFeO3 or BiFeO3 along with some impurity phases (e.g. Bi2O2CO3, Bi25FeO40 etc.).30,33,34,50 However, when RGO content in BFO–RGO was increased to 3 wt% and above, formation of an impurity phase Bi2O2CO3 along with BFO was observed (Fig. S8), (ESI†). Formation of Bi2O2CO3 impurity phase occurred might be due to the limited migration of Bi3+ and Fe3+ in the compositions having higher RGO content.50 Therefore, to obtain pure BFO nanowire-RGO nanocatalyst, we have prepared BFO–RGO nanocatalysts with 2 wt% RGO. | ||
| Fig. 1 Room temperature wide angle power XRD pattern of (a) pure GO, (b) the precipitate containing Fe(OH)3 and (Bi2O2)(OH)Cl phases, (c) BFO–RGO nanocomposite, (d) pure BiFeO3. | ||
This transformation of GO to RGO was further confirmed from the results obtained from FT-IR (Fig. 2), Raman spectroscopy (Fig. 3) and thermogravimetry analysis (TGA) (Fig. 4). EDS analysis (Fig. S9, ESI†) of BFO–RGO confirmed the presence of 98 wt% BFO in the synthesized nanocatalyst.
 | ||
| Fig. 2 FT-IR spectra of (A) BFO, (B) BFO–RGO nanocomposite and (C) GO. | ||
 | ||
| Fig. 3 Raman spectra of (A) pure BFO, (B) BFO–RGO nanocomposite and (C) GO. | ||
 | ||
| Fig. 4 TGA curve of (A) pure BFO, (B) BFO–RGO nanocomposite and (C) GO. | ||
Fig. 2 Shows FT-IR spectra of GO, pure BFO and BFO–RGO nanocomposite. It was observed that, in case of GO peaks appeared at (i) 1384 cm−1 corresponding to the stretching vibration of C–O of carboxylic group, (ii) 1720 cm−1 for carbonyl group, (iii) 1226 cm−1 for C–O stretching vibration of epoxy group (iv) 1054 cm−1 for C–O stretching vibration. This fact indicated the presence of oxygen containing functional groups (e.g. epoxy, carbonyl, carboxyl and hydroxyl) on the surface of GO. Moreover, the peak at 1621 cm−1 can be assigned to the contribution from the skeletal vibration of the graphitic domains.19,51,52 The band at 1621 cm−1 (in GO sample), which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C skeletal vibration of graphitic domains of GO.51,52 Absorption bands at the same positions were observed in FT-IR spectra of BFO–RGO nanocomposite samples. Here, carboxylic group vibration band (νCO at 1720 cm−1) was found to be disappeared and absorption intensities corresponding to C–O at 1226 and 1054 cm−1 were decreased. These results implied that, most of the oxygen containing groups of GO, particularly carboxyl groups, had been removed and some of the hydroxyl and epoxy groups were remained on the surface of RGO in BFO–RGO nanocatalyst. In FTIR spectra of pure BFO appearance of a peak at 556 cm−1, could be ascribed to lattice absorption of M–O (M = Fe3+, Bi3+), confirmed the formation of BiFeO3. Presence of this peak in BFO–RGO samples indicated the existence of BiFeO3 in BFO–RGO nanocomposite.
C skeletal vibration of graphitic domains of GO.51,52 Absorption bands at the same positions were observed in FT-IR spectra of BFO–RGO nanocomposite samples. Here, carboxylic group vibration band (νCO at 1720 cm−1) was found to be disappeared and absorption intensities corresponding to C–O at 1226 and 1054 cm−1 were decreased. These results implied that, most of the oxygen containing groups of GO, particularly carboxyl groups, had been removed and some of the hydroxyl and epoxy groups were remained on the surface of RGO in BFO–RGO nanocatalyst. In FTIR spectra of pure BFO appearance of a peak at 556 cm−1, could be ascribed to lattice absorption of M–O (M = Fe3+, Bi3+), confirmed the formation of BiFeO3. Presence of this peak in BFO–RGO samples indicated the existence of BiFeO3 in BFO–RGO nanocomposite.
Raman spectra of BFO–RGO composites (Fig. 3) also confirmed the presence of RGO in the composites. In Raman spectra of pure GO, characteristic peaks for D and G band were found at 1355 and 1597 cm−1 respectively. In case of BFO–RGO samples these peaks were appeared at 1349 and 1580 cm−1. It has been reported that Raman shift of D and G bands shifts to lower values when GO is reduced to RGO. ID/IG ratio of BFO–RGO was found to be ∼1.06 where as this ratio was ∼0.9 for pure GO.19,52 This increase of ID/IG value for BFO–RGO can be attributed to the decrease in the average size of sp2 domains upon reduction of GO during formation of BFO–RGO composite. Here also presence of peaks at 123, 153, 205, 407, 240, 256, 358, 377, 469, 528, 597, 79 and 110 cm−1 which are characteristic peaks of A1-1, A1-2, A1-3, A1-4, E-1, E-2, E-3, E-4, E-5, E-6, E-7, E-8 and E-9 modes of BFO (Fig. S10, ESI†) indicated the presence of BFO in BFO–RGO.53
TGA analysis of GO and BFO–RGO was performed in air atmosphere in the temperature range of 30–800 °C with a heating rate of 10 °C min−1. Fig. 4 shows TGA thermograms of pure GO, pure BFO and BFO–RGO nanocomposite. In TGA thermograms following points were observed: (i) pure BFO is quite stable in the temperature range of 30–800 °C. (ii) In the temperature range of 30–100 °C, GO exhibited ∼17% wt loss, which might be due to evaporation of H2O.54,55 In this temperature range ∼2% wt loss occurred for BFO–RGO. (iii) In 100–200 °C temperature range, GO showed ∼3% wt loss and a sharp weight loss occurred in the range of 200–250 °C with 15% weight loss. This was due to the removal of oxygen containing groups from GO. However, no such loss was observed for BFO–RGO. This fact clearly indicated that during synthesis of BFO–RGO, GO was converted to RGO via reduction of oxygen containing groups (e.g. carbonyl, carboxyl, epoxy groups, etc.).54,55 (iv) In 325–600 °C temperature range, the oxidative decomposition of carbon atoms of GO was observed whereas, in case of BFO–RGO, ∼2% weight loss was observed in the temperature range of 350–440 °C, which was due to the decomposition of carbon of RGO. From TGA thermogram it can be concluded that BFO–RGO nanocomposite is composed of ∼2 wt% RGO and 98 wt% BFO. Presence of an exothermic peak at 390 °C in DSC thermogram of BFO–RGO nanocatalyst indicated the decomposition of carbon in graphene at 390 °C (Fig. S11, ESI†).
Multiple point BET surface analysis of pure BFO and BFO–RGO (with 2 wt% RGO) also showed that specific surface area of BFO was 28.18 m2 g−1 where as that of BFO–RGO was 45.47 m2 g−1 (Fig. S12, ESI†). N2 adsorption–desorption isotherm of BET surface area analysis of these samples are shown in (Fig. S12, ESI†). This increased surface area is ascribed to the synergistic effects between the RGO and BFO and resulted in enhancement of catalytic activity of RGO–BFO nanocatalysts. The catalytic activities of BFO–RGO have been discussed in detail in Section 3.3., Section 3.4., Section 3.5., Section 3.6.
HRTEM and FESEM micrographs of as synthesized pure BFO and BFO–RGO nanocatalyst are shown in Fig. 5 and 6 respectively. Fig. 5A showed that BFO sample consists of BiFeO3 nanowires. Fig. 5B showed an intact, single crystalline structure of BFO nanowire. The diameter of individual nanowire was 45–200 nm and length varied from hundreds of nm to several microns. Fig. 5C and D showed the planes with interplaner spacings of 0.257 nm and 0.45 nm indicating (211) and (001) crystal faces of BiFeO3 respectively.34,40 Fig. 5E showed the nanometer thin well exfoliated sheets of RGO. Fig. 5F revealed that BFO nanowires are well dispersed on the surface of RGO sheets.
 | ||
| Fig. 5 TEM micrograph of synthesized (A) BFO, (B) individual BFO, (C) HRTEM micrograph of a typical portion of a corresponding BFO, (D) SAED pattern of an individual BFO, (E) TEM micrograph of synthesized RGO and (F) TEM micrograph of synthesized BFO–RGO nanocomposite. | ||
 | ||
| Fig. 6 FESEM images of (A) BFO and (B) BFO–RGO nanocomposite. | ||
3.2. Formation mechanism of pure BiFeO3 nanowire
The precipitate, which forms due to addition of NH4OH in the mixture of Bi(NO3)3 and FeCl3, contains small particles of Fe(OH)3 and (Bi2O2)(OH)Cl (Fig. 7A). It is important to note that, this precipitate does not contain any insoluble non-stoichiometric phases (e.g. bismuth subnitrate or bismuthyl nitrate (Bi2O3·N2O5·2H2O) etc.) which are generally formed in conventional methods employed for BiFeO3 preparation.34,35 These secondary phases create problem for single phase BiFeO3 formation.34,35 Hydrothermal treatment of Fe(OH)3 and (Bi2O2)(OH)Cl containing precipitate results in the formation of pure BiFeO3. The reactions which are involved in BiFeO3 single phase formation can be presented as follows:| 2Bi(NO3)3 + FeCl3 + 8NH4OH → (Bi2O2)(OH)Cl + Fe(OH)3 + 6NH4NO3 + 2NH4Cl + 2H2O | (3) |
| (Bi2O2)(OH)Cl + Fe(OH)3 + 2Fe(OH)3 → 2BiFeO3 + 3H2O + HCl | (4) |
 | ||
| Fig. 7 TEM images of the aliquots of reaction mixtures taken from the hydrothermal reaction of BFO after (A) 0 h, (B) 12 h and (C) 24 h. | ||
In this hydrothermal process, the growth of BFO nanowires is neither catalyst-assisted nor template directed. Based on the observation of TEM images for the aliquots of reaction mixture, collected at regular intervals of 12 h (Fig. 7), we propose that, the growth of crystals in nanowire form might proceeds via Oswald ripening process.56–58 Before hydrothermal treatment the precipitate contains nanoparticles of Fe(OH)3 and (Bi2O2)(OH)Cl (as identified by XRD (Fig. 1b) and TEM (Fig. 7A). During hydrothermal treatment, these nanoparticles undergo aggregation and rupturing (as a result of Rayleing instability56,59), followed by dissolution due to high free energy. Then the crystal growth starts from the aggregates of smaller particles. Then longer nanowires grow at the cost of smaller particles as observed by TEM (Fig. 7B) of the sample collected after 12 h of hydrothermal process. Here, the reduction of surface energy is the primary driving force for the nanowire shaped crystal growth and morphology evolution. As the reaction proceeds after 12 h, the nanoparticles start disappearing due to diffusion into growing nanowire. As a result formation of longer wires (Fig. 7C) occurs after 24 h of hydrothermal process. Similar mechanism was proposed by Lee et al. to explain the formation of BaTiO3 and SrTiO3 nanowires in a template free hydrothermal synthesis method.56
3.3. Catalytic activity of BFO–RGO nanocomposite towards reduction of 4-NP and trifluralin
To understand the effect of RGO on the catalytic activity of BFO–RGO nanocomposites, reduction reactions of 4-nitrophenol (4-NP) were performed in presence of excess NaBH4 with pure BFO (Fig. 8A) and BFO–RGO (Fig. 8B). A significant reduction of reaction completion time was observed when the reaction was catalyzed by BFO–RGO (6 min) compared to pure BFO (15 min) (Fig. 8). This enhancement of catalytic efficiency of BFO–RGO can be explained by considering following points: (i) this reduction reaction proceeds via relaying of electrons from BH4− donor to the acceptor 4-NP molecule. In aqueous medium BH4− is first absorbed on the surface of the catalyst. The hydrogen atoms, which are formed from the hydride, transfer electrons to 4-NP to reduce its functional group (e.g. –NO2 group).19–21 This electron transfer (ET) induced hydrogenation of 4-NP occurs spontaneously and BFO plays a role of storing electrons after ET from hydride. Presence of RGO not only enhances the adsorption of 4-NP onto the catalyst through π–π stacking but also facilitates the electron transfer to 4-NP via electrostatic interaction.19 This reaction followed pseudo first order kinetics and values of apparent rate constant (kapp) were 0.72 min−1 for BFO–RGO and 0.29 min−1 for pure BFO catalyzed reactions (Fig. 8D). kapp value of BFO–RGO catalyzed 4-NP reduction reaction is comparable with that of noble metal nanoparticle catalyzed reaction. This high catalytic efficiency of BFO–RGO towards reduction of –NO2 group of 4-NP provoked us to investigate the reduction of a herbicide, trifluralin, via reduction of its –NO2 groups. Trifluralin is used in various countries as a herbicide to control variety of annual grass and broadleaf weed species. But, it remains in water as herbicide residue. As it is highly toxic in nature it caused severe water pollution. BFO–RGO catalyst effectively reduced trifluralin to its colourless form, where its –NO2 groups were reduced to –NH2 groups within 25 min. | ||
| Fig. 8 Time dependent UV-vis spectral changes of the reaction mixture of 4-NP catalyzed by (A) pure BFO, (B) BFO–RGO nanocomposite and (C) trifluralin catalyzed by BFO–RGO nanocomposite in presence of excess NaBH4 and (D) pseudo first order kinetic plot of 4-NP reduction with BFO and BFO–RGO nanocomposite and trifluralin reduction catalyzed by BFO–RGO nanocomposite. | ||
As BFO–RGO nanocatalysts exhibited its enhanced catalytic efficiency towards dye degradation reaction in comparison with pure BFO, we have further explored catalytic activity of BFO–RGO towards (i) Biginelli reaction (ii) Click reaction and (iii) styrene epoxidation reaction, by performing some model reactions. When these reactions were performed in presence of pure BFO and pure RGO it was observed that no reaction occurred in presence of RGO. When pure BFO was used as catalyst, the reactions occurred to some extent but yields were significantly low in comparison with BFO–RGO catalyzed reactions.
3.4. Catalytic activity of BFO–RGO nanocomposite towards Biginelli reaction
In case of Biginelli reaction, BFO–RGO catalyzed the reaction of aldehyde, urea and dicarbonyl compounds under solventless condition very efficiently to synthesize 3,4-dihydropyrimidin-2(1H)-ones. For example, 5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-one and 5-acetyl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one were synthesized with 93% and 90% yield respectively (Table 1). When these reactions were performed for a longer time (2 h) neither increase in yield nor formation of any by product at the cost of main product was observed.
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Reaction time (min) | Yieldb (%) BFO–RGO | Yieldb (%) BFO |
| a Reaction conditions: benzaldehyde (1 mmol, 0.101 ml), urea (1.2 mmol, 72 mg), 1,3-diketone (1 mmol, 0.126 ml ethyl acetoacetate or 0.103 ml acetylacetone), BFO–RGO (25 mg), solventless condition, reaction temperature 80 °C.b Isolated yield. | |||||
| 1 | OEt |  |
30 | 93 | 65 |
| 2 | CH3 |  |
30 | 90 | 62 |

The reaction proceeds via formation of an acylimine type intermediate, from the reaction of aldehyde and urea. This intermediate forms a complex with Bi3+. The vacant d-orbital of Bi3+ promotes this complexation and stabilizes the intermediate, which intern enhances the rate of this key rate limiting step. This intermediate complex then reacts effectively with β-diketone and final product, dihydropyrimidin, forms following a cyclization and dehydration pathway.2,5 Thus BFO present in BFO–RGO acted as a catalytically active center in Biginelli reaction. The plausible mechanism of this reaction is shown in Scheme 1.
 | ||
| Scheme 1 The plausible mechanism involved in Biginelli reaction catalyzed by BFO–RGO catalyst in the synthesis of 3,4-dihydropyrimidinone. | ||
3.5. Catalytic activity of BFO–RGO nanocomposite towards Click reaction
To investigate the catalytic efficiency of BFO–RGO towards synthesis of 1,2,3-triazole via Click reaction, we have performed the three component Click reaction involving sodium azide, epoxide and non activated terminal alkynes in aqueous medium. Here also, BFO–RGO exhibited its high catalytic activity towards synthesis of 2-phenyl-2-(4-phenyl-1H-1,2,3-triazole-1-yl)ethanol (91% yield) and 2-(4-phenyl-1H-1,2,3-triazol-1-yl)cyclohexanol (89% yield) (Table 2). Performing these reactions for longer duration (∼4 h) did not affect the % of yield or formation of any other products.
|
|||||
|---|---|---|---|---|---|
| Entry | Epoxide | Triazole | Reaction time (h) | Yieldb (%) BFO–RGO | Yieldb (%) BFO |
| a Reaction conditions: alkyne (1 mmol, 0.110 ml), epoxide (1 mmol (styrene oxide 0.115 ml or, cyclohexene oxide 0.101 ml)), sodium azide (1.1 mmol, 72 mg), BFO–RGO (25 mg), water (3 ml), reaction temperature 80 °C.b Isolated yield. | |||||
| 1 |  |
 |
2 | 91 | 65 |
| 2 |  |
 |
2 | 89 | 60 |

In this reaction BFO–RGO plays a role of bifunctional catalyst. The formation of BFO-azide (to be specific Bi3+-azide (intermediate-I)) as catalytically active species helps to activate epoxide and facilitates the delivery of azide during ring opening of epoxide.9 Simultaneously another intermediate (intermediate-II) forms where acetylene coordinates with Bi3+ of BFO–RGO catalyst. This intermediate facilitates the 1,3-dipolar cycloaddition between –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– bond (intermediate-II) and azide, to produce triazole–BFO–RGO complex. Finally the protonolysis of this complex in aqueous medium results the formation of β-hydroxy-1,2,3-triazole compounds.9 The plausible mechanism of this reaction is shown in Scheme 2.
C– bond (intermediate-II) and azide, to produce triazole–BFO–RGO complex. Finally the protonolysis of this complex in aqueous medium results the formation of β-hydroxy-1,2,3-triazole compounds.9 The plausible mechanism of this reaction is shown in Scheme 2.
 | ||
| Scheme 2 The plausible reaction mechanism involved in Click reaction catalyzed by BFO–RGO catalyst for synthesis of 1,4-disubstituted 1,2,3-triazoles. | ||
3.6. Catalytic activity of BFO–RGO nanocomposite towards epoxidation of styrene
During exploration of use of BFO–RGO as catalyst for epoxidation of styrene in presence of tert-butyl hydroperoxide (TBHP), it was observed that styrene was converted to styrene oxide (Fig. S1, ESI†) with very good ∼90% selectivity and ∼79% conversion (Fig. 9). | ||
| Fig. 9 Change of conversion and product selectivity with time in the BFO–RGO catalyzed epoxidation of styrene. Reaction condition: 50 mg catalyst, 5 mmol of styrene and 12.5 mmol of TBHP were stirred in 4 ml of acetonitrile at 100 °C for 5 h. | ||
Here, TBHP first couples with BFO to form a reactive species (intermediate A). Then intermediate A further interacts with –CC– of styrene to develop a coordinated complex (intermediate B). Formation of styrene oxide from intermediate B occurs via Sharpless type mechanism.27 Here, the π–π interaction of RGO might accelerates the electron transfer between benzene ring and sp2 hybridized C-atoms of RGO,13,14,44,60 which facilitates the combination of C-atoms of vinyl groups of styrene with O-atom of TBHP and results the formation of an epoxidation product with improved conversion and selectivity. The detail mechanistic steps involved in the reaction are schematized in Scheme 3.
 | ||
| Scheme 3 The plausible mechanism involved in the BFO–RGO catalyzed synthesis of styrene oxide from styrene in presence of TBHP. | ||
However, when the reaction mixture was allowed to react for more than 5 h it was observed that benzaldehyde was forming due to the reaction of styrene oxide and TBHP (Fig. S1, ESI†). Formation of benzaldehyde proceeds via following mechanism17 (Scheme 4):
 | ||
| Scheme 4 The plausible reaction mechanism involved in the formation of benzaldehyde due to the reaction of styrene oxide and TBHP. | ||
The conversion and product selectivity were calculated as follows:
 | (5) |
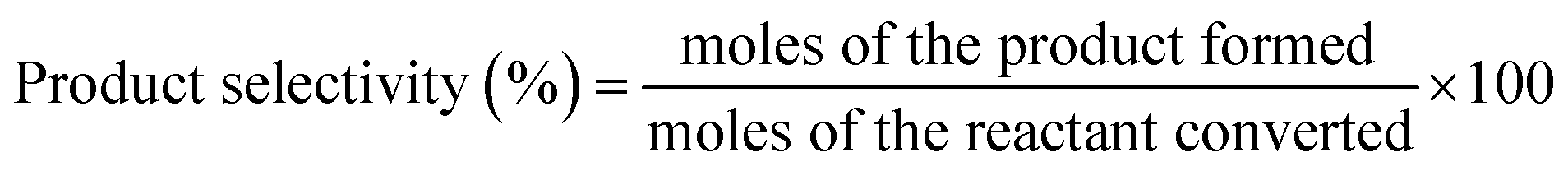 | (6) |
The presence of BFO introduces the magnetic character in BFO–RGO nanocatalyst (saturation magnetization (Ms) = 2.69 emu g−1 and coercivity (Hc) = 1.145 Oe). The super paramagnetic character of this catalyst allows their easy magnetic separation from the reaction mixture with an external magnet (Fig. 10).
 | ||
| Fig. 10 Room temperature magnetic hysteresis loop of (A) BFO–RGO nanocomposite, inset (B) shows the decolourization of 4-NP in presence of NaBH4 and BFO–RGO catalyst and magnetic separation of BFO–RGO catalyst by applying a magnet externally after completion of reaction. | ||
3.7. Reusability of magnetically separable BFO–RGO catalyst after catalysis reactions
After completion of the reactions the catalyst, BFO–RGO, was magnetically separated, washed with water several times and dried. The recovered catalyst was again used for all reactions. It was observed that activity of the catalyst remained almost same up to 5 cycles slight decrease of catalytic activity for Biginelli, Click reaction and epoxidation reaction was observed after 7th reactions (∼2% decrease). For 4-nitro phenol reaction, BFO–RGO exhibited good activity after 5 cycles, with 100% conversion within 6–7 min. After 8 cycles of reactions, the reaction time observed for 100% conversion was within 10–12 min. (Fig. S13–S16, ESI†). XRD patterns and FESEM image of the reused catalyst also showed no significant changes in their crystal structure as well as morphology compared to the fresh catalyst (Fig. 11A and B). | ||
| Fig. 11 (A) XRD and (B) FESEM image of the recycled BFO RGO catalyst. | ||
4. Conclusions
The highlight of this paper is that, here we are reporting for the first time a synthetic methodology for preparation of BiFeO3 nanowire-RGO nanocatalyst. The synthesized BFO–RGO nanocatalyst consists of pure single phase BiFeO3 nanowires, which are embedded on the surface of nanometer thin RGO sheets. To the best of our knowledge, this versatile catalytic activity of BiFeO3–RGO nanocatalyst towards (i) reduction of 4-nitrophenol (4-NP) and a herbicide, trifluralin in presence of NaBH4 (ii) Biginelli reaction (iii) epoxidation of styrene and (iv) Click reactions has been reported here for the first time.The excellent catalytic activity of this catalyst arises from the synergistic effect of RGO and BFO. It is important to note that, as Biginelli reaction can be performed in solventless condition and Click reaction in aqueous medium, this catalyst promotes the environmentally green approach to conduct these organic synthesis reactions. The usefulness of BFO–RGO as catalyst for various important organic reactions, its high catalytic efficiency, easy separation and good reusability make it an attractive nanocatalyst.
Acknowledgements
Dr N. N. Ghosh gratefully acknowledges financial support from CSIR, India (CSIR Sanction letter No. 02(147)/13/EMR-II).Notes and references
- H. G. Alvim, T. B. Lima, A. L. de Oliveira, H. C. de Oliveira, F. M. Silva, F. C. Gozzo, R. Y. Souza, W. A. da Silva and B. A. Neto, J. Org. Chem., 2014, 79, 3383 CrossRef CAS PubMed
.
- B. K. Banik, A. T. Reddy, A. Datta and C. Mukhopadhyay, Tetrahedron Lett., 2007, 48, 7392 CrossRef CAS
- J. Mondal, T. Sen and A. Bhaumik, Dalton Trans., 2012, 41, 6173 RSC
- J. Javidi, M. Esmaeilpour and F. N. Dodeji, RSC Adv., 2015, 5, 308 RSC
- K. Ramalinga, P. Vijayalakshmi and T. Kaimal, Synlett, 2001, 6, 0863 CrossRef
- A. Debache, M. Amimour, A. Belfaitah, S. Rhouati and B. Carboni, Tetrahedron Lett., 2008, 49, 6119 CrossRef CAS
- S. L. Jain, S. Singhal and B. Sain, Green Chem., 2007, 9, 740 RSC
- A. N. Prasad, B. Thirupathi, G. Raju, R. Srinivas and B. M. Reddy, Catal. Sci. Technol., 2012, 2, 1264 CAS
- H. Naeimi and V. Nejadshafiee, New J. Chem., 2014, 38, 5429 RSC
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS PubMed
- Z. Li, S. Wu, D. Zheng, H. Ding, X. Wang, X. Yang, Q. Huo, J. Guan and Q. Kan, ChemPlusChem, 2014, 79, 716 CrossRef CAS
- X. Zhang, G. Wang, M. Yang, Y. Luan, W. Dong, R. Dang, H. Gao and J. Yu, Catal. Sci. Technol., 2014, 4, 3082 CAS
- M. Liu, X. Wang, Y. Chen and L. Dai, RSC Adv., 2015, 5, 61481 RSC
- G. Bian, P. Jiang, H. Zhao, K. Jiang, L. Hu, Y. Dong and W. Zhang, ChemistrySelect, 2016, 1, 1384 CrossRef CAS
- V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2006, 8, 689 RSC
- Z.-Q. Shi, Z.-P. Dong, J. Sun, F.-W. Zhang, H.-L. Yang, J.-H. Zhou, X.-H. Zhu and R. Li, Chem. Eng. J., 2014, 237, 81 CrossRef CAS
- A. Wang and H. Jing, Dalton Trans., 2014, 43, 1011 RSC
- D. Yang, N. Yang and J. Ge, CrystEngComm, 2013, 15, 7230 RSC
- D. Moitra, B. Ghosh, M. Chandel, R. Jani, M. Patra, S. Vadera and N. Ghosh, RSC Adv., 2016, 6, 14090 RSC
- H. Hu, J. H. Xin, H. Hu, X. Wang, D. Miao and Y. Liu, J. Mater. Chem. A, 2015, 3, 11157 CAS
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410 RSC
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1 CrossRef CAS
- T. Sun, Z. Zhang, J. Xiao, C. Chen, F. Xiao, S. Wang and Y. Liu, Sci. Rep., 2013, 3, 2527 Search PubMed
- Z. Zhang, F. Xiao, J. Xi, T. Sun, S. Xiao, H. Wang, S. Wang and Y. Liu, Sci. Rep., 2014, 4, 4053 Search PubMed
- Z. Zhang, T. Sun, C. Chen, F. Xiao, Z. Gong and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 21035 CAS
- B. T. Worrell, S. P. Ellery and V. V. Fokin, Angew. Chem., Int. Ed., 2013, 52, 13037 CrossRef CAS PubMed
- M. L. Kuznetsov, B. G. Rocha, A. J. Pombeiro and G. B. Shul'pin, ACS Catal., 2015, 5, 3823 CrossRef CAS
- Y. Q. Kang, M.-S. Cao, J. Yuan and X. L. Shi, Mater. Lett., 2009, 63, 1344 CrossRef CAS
- Y. Wang, L. Zhou, M. Zhang, X. Chen, J. Liu and Z. Liu, Appl. Phys. Lett., 2004, 84, 1731 CrossRef CAS
- E. Aguiar, M. Ramirez, F. Moura, J. A. Varela, E. Longo and A. Simoes, Ceram. Int., 2013, 39, 13 CrossRef CAS
- S. Ghosh, S. Dasgupta, A. Sen and H. Sekhar Maiti, J. Am. Ceram. Soc., 2005, 88, 1349 CrossRef CAS
- Y. Li, H.-J. Yang, W.-G. Yang, Z.-L. Hou, J.-B. Li, H.-B. Jin, J. Yuan and M.-S. Cao, Mater. Lett., 2013, 111, 130 CrossRef CAS
- Y. Wang, G. Xu, L. Yang, Z. Ren, X. Wei, W. Weng, P. Du, G. Shen and G. Han, J. Am. Ceram. Soc., 2007, 90, 3673 CrossRef CAS
- B. Liu, B. Hu and Z. Du, Chem. Commun., 2011, 47, 8166 RSC
- M. Popa, D. Crespo, J. M. Calderon-Moreno, S. Preda and V. Fruth, J. Am. Ceram. Soc., 2007, 90, 2723 CrossRef CAS
- F. Gao, Y. Yuan, K. Wang, X. Chen, F. Chen, J. Liu and Z. Ren, Appl. Phys. Lett., 2006, 89, 102506 CrossRef
- T.-J. Park, Y. Mao and S. S. Wong, Chem. Commun., 2004, 2708 RSC
- S. Xie, J. Li, R. Proksch, Y. Liu, Y. Zhou, Y. Liu, Y. Ou, L. Lan and Y. Qiao, Appl. Phys. Lett., 2008, 93, 222904 CrossRef
- L. Zhang, X.-F. Cao, Y.-L. Ma, X.-T. Chen and Z.-L. Xue, J. Solid State Chem., 2010, 183, 1761 CrossRef CAS
- S. Li, J. Zhang, M. G. Kibria, Z. Mi, M. Chaker, D. Ma, R. Nechache and F. Rosei, Chem. Commun., 2013, 49, 5856 RSC
- A. S. Nia, S. Rana, D. Döhler, X. Noirfalise, A. Belfiore and W. H. Binder, Chem. Commun., 2014, 50, 15374 RSC
- N. Salam, A. Sinha, A. S. Roy, P. Mondal, N. R. Jana and S. M. Islam, RSC Adv., 2014, 4, 10001 RSC
- Z. Li, Y. Shen, Y. Guan, Y. Hu, Y. Lin and C.-W. Nan, J. Mater. Chem. A, 2014, 2, 1967 CAS
- Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, G. Hu, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124 CrossRef CAS PubMed
- Y. Gao, X. Chen, J. Zhang, H. Asakura, T. Tanaka, K. Teramura, D. Ma and N. Yan, Adv. Mater., 2015, 27, 4688 CrossRef CAS PubMed
- H. Hong, L. Hu, M. Li, J. Zheng, X. Sun, X. Lu, X. Cao, J. Lu and H. Gu, Chem.–Eur. J., 2011, 17, 8726 CrossRef CAS PubMed
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef
- B. Naik, S. Hazra, V. S. Prasad and N. N. Ghosh, Catal. Commun., 2011, 12, 1104 CrossRef CAS
- B. Naik, S. Hazra, D. Desagani, B. K. Ghosh, M. K. Patra, S. R. Vadera and N. N. Ghosh, RSC Adv., 2015, 5, 40193 RSC
- Z. Li, Y. Shen, C. Yang, Y. Lei, Y. Guan, Y. Lin, D. Liu and C.-W. Nan, J. Mater. Chem. A, 2013, 1, 823 CAS
- G.-S. Wang, Y. Wu, Y.-Z. Wei, X.-J. Zhang, Y. Li, L.-D. Li, B. Wen, P.-G. Yin, L. Guo and M.-S. Cao, ChemPlusChem, 2014, 79, 375 CrossRef CAS
- M. Fu, Q. Jiao, Y. Zhao and H. Li, J. Mater. Chem. A, 2014, 2, 735 CAS
- R. Das, G. G. Khan, S. Varma, G. D. Mukherjee and K. Mandal, J. Phys. Chem. C, 2013, 117, 20209 CAS
- D. Moitra, M. Chandel, B. K. Ghosh, R. K. Jani, M. K. Patra, S. R. Vadera and N. N. Ghosh, RSC Adv., 2016, 6, 76759 RSC
- M. Zong, Y. Huang, Y. Zhao, X. Sun, C. Qu, D. Luo and J. Zheng, RSC Adv., 2013, 3, 23638 RSC
- U. A. Joshi and J. S. Lee, Small, 2005, 1, 1172 CrossRef CAS PubMed
- S. H. Yu, B. Liu, M. S. Mo, J. H. Huang, X. M. Liu and Y. T. Qian, Adv. Funct. Mater., 2003, 13, 639 CrossRef CAS
- W. Wang, C. Xu, G. Wang, Y. Liu and C. Zheng, Adv. Mater., 2002, 14, 837 CrossRef CAS
- D. Quéré, J.-M. Di Meglio and F. Brochard-Wyart, Science, 1990, 249, 1256 Search PubMed
- J. An, L. Zhu, N. Wang, Z. Song, Z. Yang, D. Du and H. Tang, Chem. Eng. J., 2013, 219, 225 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of spectral data of the synthesized compounds obtained from Biginelli, Click reaction and styrene epoxidation are shown in (Fig. S1–S7), Fig. S8 room temperature wide angle powder XRD patterns of 97BFO–3RGO (3 wt% RGO content BiFeO3 nanowire), Fig. S9 EDS spectra of synthesized BFO–RGO nanocatalyst, Fig. S10 Raman spectra of pure BFO, Fig. S11 TGA and DSC thermogram of BFO–RGO nanocatalyst, Fig. S12 N2 adsorption–desorption isotherm of BFO and BFO–RGO nanocatalyst, Fig. S13–S16 reusability of magnetically separable BFO–RGO catalyst after catalysis reactions. See DOI: 10.1039/c6ra22077k |
| This journal is © The Royal Society of Chemistry 2016 |