
Photodegradation behavior and mechanism of poly(ethylene glycol-co-1,4-cyclohexanedimethanol terephthalate) (PETG) random copolymers: correlation with copolymer composition
Abstract
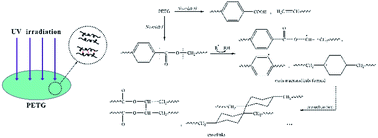
The effect of copolymer composition on the photodegradation behavior and the mechanism of poly(ethylene glycol-co-1,4-cyclohexanedimethanol terephthalate) (PETG) random copolymers with different 1,4-cyclohexanedimethanol (CHDM) content were first investigated. The changes in surface chemical groups of the PETG copolymers after UV irradiation were characterized by X-ray photoelectron spectroscopy (XPS) and attenuated total reflectance Fourier transform-infrared (ATR-FTIR) spectroscopy. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were used to probe the thermal properties of the PETG copolymers before and after UV irradiation. The crosslinking degree of the PETG copolymers after UV irradiation was evaluated by gel content measurement. The photooxidation rate of the PETG copolymers increased with increasing CHDM content. Namely, the inherent photostability of the PETG copolymers decreased with increasing CHDM content. The PETG copolymers with different compositions exhibited the similar photooxidation mechanism. The presence of CHDM in the PETG molecular chains accelerated the formation of photoproducts. The photoproducts of the PETG copolymers were consisted of aliphatic alcohol, anhydride, benzoic acid, double bond and aliphatic acid as end-groups and molecular terephthalic acid. Moreover, the crosslinking products formed during UV irradiation were not further oxidized in the whole irradiation period (0–800 h). The glass transition temperatures (Tgs) of the PETG copolymers after UV irradiation increased due to the irradiation crosslinking. The increment of Tg increased gradually with increasing CHDM content. Therefore, the higher the CHDM content was, the higher the crosslinking degree obtained.
 Please wait while we load your content...
Please wait while we load your content...

