
Solution blending preparation of polycarbonate/graphene composite: boosting the mechanical and electrical properties†
Abstract
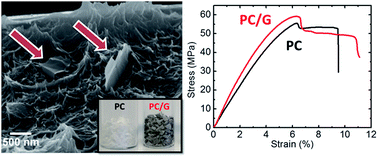
We report on the preparation of polycarbonate-based graphene (PC/G) composites, by using a simple and scalable solution blending method to disperse single- (SLG) and few-layer (FLG) graphene flakes, prepared by liquid phase exfoliation (LPE), in the polymer matrix. A solvent-exchange process is carried out to re-disperse the exfoliated SLG/FLG flakes in an environmentally friendly solvent, i.e. 1,3-dioxolane, which is also used to dissolve the polycarbonate pellets, thus facilitating the mixing of the polymer dispersion with the SLG/FLG flakes. The loading of SLG/FLG flakes improves the mechanical and thermal properties, as well as the electrical conductivity of the polymer, reaching a +26% improvement of the elastic modulus at 1 wt% loading, and an electrical conductivity 10−3 S m−1 at 10 wt% with a percolation threshold achieved at 0.55 vol%. The as-prepared PC/G composite with the aforementioned reinforced properties can be a promising material for 3D printing-based applications.
 Please wait while we load your content...
Please wait while we load your content...

