
Mild synthesis of mercaptonitriles from vinyl nitriles and their cyclization reactions†
Philip Caspariab,
Frank A. Nüeschab,
Antonia Neelsc and
Dorina M. Opris*a
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Functional Polymers, Ueberlandstr. 129, CH-8600, Dübendorf, Switzerland. E-mail: dorina.opris@empa.ch
bEcole Polytechnique Fédérale de Lausanne (EPFL), Institut des Matériaux, Station 12, CH 1015, Lausanne, Switzerland
cEmpa, Swiss Federal Laboratories for Materials Science and Technology, Center for X-ray Analytics, Ueberlandstr. 129, CH-8600, Dübendorf, Switzerland
First published on 6th October 2016
Abstract
Thiol–ene addition of thioacetic acid A is widely used in the synthesis of thiols from vinyl precursors, but so far has not been conducted on non-conjugated vinyl nitriles. The challenge when vinyl nitriles are used is to selectively conduct the thiol–ene addition, while avoiding the nucleophilic addition of A to the nitrile group. We have found that vinyl nitriles give selective UV-induced thiol–ene addition in the presence of photoinitiators as long as a stoichiometric amount of A to the vinyl group and sterically unhindered vinyls are used. In contrast, when a sterically hindered vinyl is used, the nucleophilic addition of the nitrile is favoured. The prepared mercaptonitriles can easily undergo cyclization reactions in basic and acidic conditions as well as in the presence of silica gel. This illustrates the high reactivity of nitriles towards thiol addition. 1,2-Ethanedithiol B is presented as an alternative reagent to A as it allows conversion of vinyl nitriles directly into mercaptonitriles under mild and non-acidic reaction conditions.
Introduction
The radical addition of thiols to carbon–carbon double bonds is widely known as the thiol–ene reaction. This reaction is regioselective and gives the anti-Markovnikov adduct as the major product.1 Due to high conversion, small amounts of by-products and fast reaction times, the thiol–ene reaction is often referred to as “thiol click chemistry” in the literature.2–5 The addition is mediated by either thermal6 or by UV radical initiators. The thiol–ene reaction is a versatile tool in biochemistry7–10 and polymer science. In polymer science, the thiol–ene reaction is a well-established process for the preparation of dithiol monomers6 and in the synthesis of polythioethers11–13 and polythioesters.14 Due to its high conversion it has often been used in post-polymerization modifications.15–21 When photoinitiators are used, the reaction proceeds to completion within few minutes.7 In their absence thiols can be activated by UV irradiation but the reaction rate is considerably lower.8 The addition of thioacetic acid A to alkenes followed by acetyl cleavage is one of the most frequently used synthetic routes to thiols. Cleavage of the acetyl moiety can be conducted under acidic6 and basic conditions22 or by reduction with lithium aluminium hydride.23 Hydrogen sulfide addition to alkenes leads to formation of significant amount of dialkylsulfides side-products. Already in 1941 Schjanberg et al. published the selective thiol–ene addition of A to non-conjugated pentenoic acids.24 Later, Brown et al. reported the selective thiol–ene addition of A to allyl alcohol, allyl acetate, and maleic anhydride mediated by peroxides.25 More recently thiol–ene addition of non-conjugated vinyl amides using BEt3 and O2 as initiator was reported.26 Thiol–ene addition of aliphatic thiols to allyl cyanide is known,27,28 however, the formed sulfides cannot be converted into mercaptonitriles. While selective 1,4-addition of A to acrylonitrile under basic conditions is known,29 the thiol–ene addition of A and 1,2-ethanedithiol B to non-conjugated vinyl nitrile compounds i.e. allyl cyanide was not explored so far. The addition of A to aliphatic nitriles was reported to occur under acidic or basic conditions with formation of thioamides.30,31 It is also known that mercaptonitriles can undergo intramolecular reactions under acidic conditions.32 Given the reactivity of A to nitriles it was unclear whether a selective thiol–ene addition of A to various vinyl nitriles followed by the deprotection of the thioester to thiol can be realized.In polymer science, mercaptonitriles are attractive precursor for the functionalization of polyvinylsiloxanes via thiol–ene reaction.17 We have recently shown that the dielectric permittivity (ε′) of polysiloxanes modified with polar nitrile groups increases linearly with the content of the nitrile groups. By increasing the content of nitrile groups on the polysiloxane, it is to be expected that the permittivity can be further increased. To be attractive precursors for post-polymerization reactions, mercaptonitriles must be synthesized cost-efficiently on g-scale and have to be stable during thiol–ene addition to the polyvinylsiloxane.
This work describes the thiol–ene reaction of A and 1,2-ethanedithiol B to allyl cyanide, allyl malononitrile, and 3-vinyl-1,3,5-tricarbopentanitrile which should allow formation of thiols with different content of nitrile groups in two steps or directly, respectively.
Experimental section
General remarks
All reactions were carried out in dried glassware under argon atmosphere. Unless otherwise stated, all chemicals were reagent grade and used as received. Methanol, dichloromethane (DCM), heptane, ethyl acetate, petroleum ether and tetrahydrofurane (THF) were purchased from VWR. Anhydrous methanol was purchased from Sigma Aldrich. Silica gel was purchased from VWR (‘Normasil 60’ 40–63 μm). Analytical TLC was carried out on ‘TLC Silica gel 60 F254’ from Merck. TLC plates were visualized with UV light or aqueous KMnO4 solution. THF was dried over sodium and distilled prior use. All other chemicals were purchased from Sigma-Aldrich. A mercury vapour UV-light source UVAHAND 250 GSH1 from Dr Hönle AG without additional filters providing an irradiation intensity of 15 mW cm−2 in the frequency range between 320 and 600 nm was used. 1H and 13C NMR spectra were recorded on a Bruker Avance III 400 NMR spectrometer using a 5 mm BBO Prodigy™ CryoProbe at 400.18 and 100.63 MHz, respectively. Chemical shifts (δ) in ppm are calibrated to residual solvent peaks according to literature.33 Mass spectrometry was conducted by the laboratory of organic chemistry of ETH Zurich. EI-MS was measured with Waters' AutoSpec Ultima (EI-triSector-MS), ESI-MS with Bruker's maXis (ESI/NanoSpray-Qq-TOF-MS). Elemental analysis was determined with LECO TruSpec Micro, LECO RO-478 and LECO CHNS-932 by the laboratory of organic chemistry of ETH Zurich. For the broadband dielectric spectroscopy (BDS) measurements a high impedance Alpha Analyzer combined with a Quatro temperature controller (both from Novocontrol) has been employed to cover a broad frequency from 0.1 Hz to 1 MHz. Two stainless steel discs with a diameter of 20 mm served as electrodes which were separated by three glass fibers with a diameter of 100 μm.![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2–CH2–), 2.42 (t, 3J = 8.0 Hz, 2H, –CH2–C2–CN), 2.35 (s, 3H, C3–CO–S–), 1.94 (q, 3J = 7.1 Hz, 2H, –CH2–C2–CH2–CN). 13C NMR (100 MHz, CDCl3) δ, ppm: 195.33 (CH3–
2–CH2–), 2.42 (t, 3J = 8.0 Hz, 2H, –CH2–C2–CN), 2.35 (s, 3H, C3–CO–S–), 1.94 (q, 3J = 7.1 Hz, 2H, –CH2–C2–CH2–CN). 13C NMR (100 MHz, CDCl3) δ, ppm: 195.33 (CH3–![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) O–S–), 119.09 (–CH2–CH2–N), 30.77 (H3–CO–S–), 28.00 (–S–H2–CH2–), 25.93 (–S–CH2–H2–), 16.42 (–CH2–H2–CN). MS (EI) m/z (%): 43.02 (100), 128.02 (10.97), 143.04 (4.38). EA: calculated: [C] 50.32% [H] 6.33% [N] 9.78% [O] 11.17 [S] 22.39 found: [C] 50.27% [H] 6.54% [N] 9.54% [O] 11.40 [S] 22.22%.2–CH2–), 2.54 (t, 3J = 7.0 Hz, 2H, –CH2–C2–CN), 1.96 (q, 3J = 7.0 Hz, 2H, –CH2–C2–CH2–), 1.38 (t, 3J = 8.3 Hz, 2H, S–CH2–). 13C NMR (100 MHz, CDCl3) δ, ppm: 119.10 (–CH2–CH2–N), 29.39 (–S–H2–CH2–), 23.34 (–S–CH2–H2–), 15.90 (–CH2–H2–CN). MS (EI) m/z (%): 54.03 (100), 68.05 (23.84), 101.03 (97.52).
O–S–), 119.09 (–CH2–CH2–N), 30.77 (H3–CO–S–), 28.00 (–S–H2–CH2–), 25.93 (–S–CH2–H2–), 16.42 (–CH2–H2–CN). MS (EI) m/z (%): 43.02 (100), 128.02 (10.97), 143.04 (4.38). EA: calculated: [C] 50.32% [H] 6.33% [N] 9.78% [O] 11.17 [S] 22.39 found: [C] 50.27% [H] 6.54% [N] 9.54% [O] 11.40 [S] 22.22%.2–CH2–), 2.54 (t, 3J = 7.0 Hz, 2H, –CH2–C2–CN), 1.96 (q, 3J = 7.0 Hz, 2H, –CH2–C2–CH2–), 1.38 (t, 3J = 8.3 Hz, 2H, S–CH2–). 13C NMR (100 MHz, CDCl3) δ, ppm: 119.10 (–CH2–CH2–N), 29.39 (–S–H2–CH2–), 23.34 (–S–CH2–H2–), 15.90 (–CH2–H2–CN). MS (EI) m/z (%): 54.03 (100), 68.05 (23.84), 101.03 (97.52).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N2), 3.51 (t, 3J = 6.6 Hz, 2H, –CH2–C2–S–), 3.18 (t, 3J = 7.1 Hz, 2H, –C(NH2)–C2–CH2–), 2.56 (q, 3J = 6.8 Hz, 2H, –CH2–C2–CH2–). 13C NMR (100 MHz, DMSO-d6) δ, ppm: 203.78 (–S–NH2), 39.38 (–S–H2–CH2–), 37.61 (–C(NH2)–H2–CH2–), 27.55 (–CH2–H2–CH2–).
N2), 3.51 (t, 3J = 6.6 Hz, 2H, –CH2–C2–S–), 3.18 (t, 3J = 7.1 Hz, 2H, –C(NH2)–C2–CH2–), 2.56 (q, 3J = 6.8 Hz, 2H, –CH2–C2–CH2–). 13C NMR (100 MHz, DMSO-d6) δ, ppm: 203.78 (–S–NH2), 39.38 (–S–H2–CH2–), 37.61 (–C(NH2)–H2–CH2–), 27.55 (–CH2–H2–CH2–).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 5 (35 g, 35%) as colourless viscous liquid. RF = 0.4 (heptane/ethyl acetate 2:1). 1H NMR (400 MHz, CDCl3) δ, ppm: 5.91–5.80 (m, 1H, –CH2–CCH2), 5.43–5.38 (m, 2H, –CH2–CHC2), 3.78 (t, 3J = 6 Hz, 1H, (NC)2–C–CH2–), 2.75 (dt, 4J = 1 Hz, 3J = 6.6 Hz 2H, CH–C2–CH). 13C NMR (100 MHz, CDCl3) δ, ppm: 129.29 (–HCH2), 122.55 (–CHH2), 112.35 (–CN), 34.74 (–CH–H2–), 23.16 (–H–CH2–).:1, RF = 0.3). 6 was obtained as colourless viscous liquid (1.35 g, 74%). 1H NMR (400 MHz, CDCl3) δ, ppm: 3.87 (t, 3J = 6.8 Hz, 1H, (NC)2–C–CH2–), 2.95 (t, 3J = 6.8 Hz, 2H, CH2–C2–SAc), 2.36 (s, 3H, CH2–S–C3), 2.13–2.06 (m, 2H, CH–C2–CH2–), 1.95–1.84 (m, 2H, CH–CH2–C2–). 13C NMR (100 MHz, CDCl3) δ, ppm: 195.65 (–S–O–CH3), 112.53 (–N), 30.76 (–S–C(O)–H3), 29.55 (CH2–H2–CH2), 27.22 (H2–SAc), 26.84 (CH–H2–), 22.30 (–H–CH2–). MS (EI+) m/z (%): 182.05 (5.53), 43.02 (100). EA: calculated: [C] 52.73% [H] 5.53% [N] 15.37% [O] 8.78% [S] 17.59% found: [C] 52.53% [H] 5.75% [N] 15.31% [O] 9.05 [S] 17.53%.–CH2–), 2.61–2.67 (m, 2H, CH2–C2–SH), 2.23–2.17 (m, 2H, CH–C2–CH2–), 1.98–1.90 (m, 2H, CH–CH2–C2–), 1.43 (t, 3J = 8.1 Hz, 1H, –CH2–S). 13C NMR (100 MHz, CDCl3) δ, ppm: 195.60 (–S–O–CH3), 112.54 (–N), 29.47 (CH2–H2–CH2), 30.39 (H2–SH), 23.39 (CH–CH2–), 22.46 (–CH–H2–). HRMS (ESI): calc. C6H9N2S m/z: 141.0481 [M + H]; found 141.0481 [M + H].:1). 1H NMR (400 MHz, CDCl3) δ, ppm: 4.33 (br s, 2H, –NH2), 2.94 (t, 3J = 5.6 Hz, 2H, –CH2–C2–S), 2.35 (t, 3J = 6.4 Hz, 2H, –C2–CH2–CH2–), 2.03–1.96 (m, 2H, –CH2–C2–CH2). 13C NMR (100 MHz, CDCl3) δ, ppm: 154.19 (C(–NH2)–S), 120.13 (–N), 72.94 ((NC)–C–S), 28.17 (S–H2–CH2–CH2–), 24.37 (S–CH2–CH2–H2–), 22.47 (–CH2–H2–CH2–). HRMS (ESI) calc. for C6H9N2S [M + H] 141.0481, found 141.0481. EA: calculated: [C] 51.40% [H] 5.74% [N] 19.98% found: [C] 51.44% [H] 5.83% [N] 19.73%.:1) to give a pale yellowish solid (9.0 g). The solid was dissolved in ethyl acetate and precipitated by slow addition n-heptane to obtain 9 (8.2 g, 31%) as colourless solid. RF = 0.5 (heptane/ethyl acetate 1:1). 1H NMR (400 MHz, CDCl3) δ, ppm: 5.67 (d, 3J = 16 Hz, 1H, CHCH–), 5.58 (d, 3J = 11 Hz, 1H, CHCH–), 5.44 (dd, 3J = 10.2 Hz, 3J = 16.8 Hz, 1H, CH2C–), 2.57–2.39 (m, 4H, CH2–C2–CN), 2.24–2.17 (m, 2H, CH–CH2–CN), 2.00–1.92 (m, 2H, CH–CH2–CN). 13C NMR (100 MHz, CDCl3) δ, ppm: 132.76 (CH2H–), 122.17 (H2CH–), 117.98 (Cquart–N), 117.85 (CH2–N), 45.81 (Cquart), 34.22 (H2–CH2CN), 13.66 (CH2–H2–CN). MS (EI) m/z (%): 55.05 (49.53), 80.05 (34.74), 92.05 (70.69), 119.06 (100). EA: calculated: [C] 69.34% [H] 6.40% [N] 24.26% found: [C] 69.21% [H] 6.33% [N] 24.16%.:1) to give (0.4 g, 58%) of 12. RF = 0.2 (heptane/ethyl acetate 1:1). 1H NMR (400 MHz, acetone-d6) δ, ppm: 8.68 (br s, 4H, –CS–N2), 5.71 (dd, 3J = 10 Hz, 3J = 17 Hz, 1H, CH2CH–), 5.42 (d, 3J = 17 Hz, 1H, CHCH–), 5.35 (d, 3J = 10 Hz, 1H, CHCH–), 2.77–2.63 (m, 4H, CH2–C2–C(S)NH2), 2.28–2.11 (m, 4H, C2–CH2–CN). 13C NMR (100 MHz, acetone-d6) δ, ppm: 208.12 (–(S)NH2), 136.79 (CH2H–), 120.21 (H2CH–), 117.37 (Cquart–N), 45.60 (Cquart), 40.14 (H2–H2C(S)NH2), 37.31 (CH2–H2–C(S)NH2); MS (ESI): calc. for C10H16N3S2 [M + H] 242.0780; found 242.0779.2–C2–SH), 2.68 (t, 3J = 7.2 Hz, 2H, –S–C2–CH2–), 2.51 (t, 2H, –CH2–C2–CN, J = 6.8 Hz), 1.94 (dt, 4J = 7.44 Hz, 2H, –CH2–C2–CH2–CN), 1.71 (t, 3J = 8.1 Hz, 1H, –CH2–S). 13C NMR (100 MHz, CDCl3) δ, ppm: 119.27 (CH2–N), 36.20 (HS–CH2–H2–S–), 30.66 (–S–H2–CH2–), 25.36 (–CH2–H2–CH2–), 24.77 (HS–H2–CH2–), 16.14 (–CH2–H2–CN). MS (EI+) m/z (%): 114.03 (94.2), 161.03 (60.5). EA: calculated: [C] 44.68% [H] 6.87% [N] 8.68% [S] 39.76 found: [C] 44.51% [H] 7.12% [N] 8.58% [S] 39.78.–CH2–), 2.79–2.69 (m, 4H, –S–(C2)2–SH), 2.64 (t, 3J = 7.1 Hz, 2H, CH2–C2–S–), 2.21–2.15 (m, 2H, CH–C2–CH2), 1.96–1.88 (m, 2H, CH2–C2–CH2), 1.72 (t, 3J = 8.0 Hz, 1H, CH2–S–). 13C NMR (100 MHz, CDCl3) δ, ppm: 112.47 ((N)2–CH–), 36.11 (–H2–CH2–SH), 30.62 (–H2–CH2–S–), 29.64 (CH–H2–CH2), 26.11 (CH2–H2–CH2), 24.72 (CH2–H2–SH), 22.48 ((CN)2–H–). MS (ESI) m/z (%): 167 (100), 359.24 (22.5), 423 (23.8). EA: calculated: [C] 47.97% [H] 6.04% [N] 13.98% [S] 32.01%/found: [C] 47.90% [H] 6.24% [N] 13.74% [S] 31.86%.:1) to give (0.09 g, 34%) 15 as yellowish liquid. 1H NMR (400 MHz, CDCl3) δ, ppm: 2.83–2.72 (m, 4H, HS–C2–C2–S), 2.67 (t, 2H, –S–C2–CH2–Cquart, 3J = 8 Hz), 2.57 (t, 3J = 8 Hz, 4H, –CH2–C2–CN), 2.07 (t, 3J = 8 Hz, 4H, –C2–CH2–CN), 1.96 (t, 3J = 8 Hz, 2H, –C2–Cquart), 1.73 (t, 1H, –S). 13C NMR (100 MHz, CDCl3) δ, ppm: 119.96 (Cquart–N), 117.89 (–CH2–N), 40.62 (quart), 36.65 (–H2–SH), 35.78 (S–CH2–H2–Cquart–), 31.63 (H2–CH2–CN), 26.79 (S–H2–CH2–Cquart–), 24.89 (HS–CH2–H2–S), 13.48 (H2–CN). MS (EI+) m/z (%): 61.01 (100), 69.05 (15.55), 207.08 (17.18), 267.08 (13.02). EA: calculated: [C] 53.90% [H] 6.41% [N] 15.71% [S] 23.98 found: [C] 53.78% [H] 6.60% [N] 15.43% [S] 23.82%.
:1) to give 5 (35 g, 35%) as colourless viscous liquid. RF = 0.4 (heptane/ethyl acetate 2:1). 1H NMR (400 MHz, CDCl3) δ, ppm: 5.91–5.80 (m, 1H, –CH2–CCH2), 5.43–5.38 (m, 2H, –CH2–CHC2), 3.78 (t, 3J = 6 Hz, 1H, (NC)2–C–CH2–), 2.75 (dt, 4J = 1 Hz, 3J = 6.6 Hz 2H, CH–C2–CH). 13C NMR (100 MHz, CDCl3) δ, ppm: 129.29 (–HCH2), 122.55 (–CHH2), 112.35 (–CN), 34.74 (–CH–H2–), 23.16 (–H–CH2–).:1, RF = 0.3). 6 was obtained as colourless viscous liquid (1.35 g, 74%). 1H NMR (400 MHz, CDCl3) δ, ppm: 3.87 (t, 3J = 6.8 Hz, 1H, (NC)2–C–CH2–), 2.95 (t, 3J = 6.8 Hz, 2H, CH2–C2–SAc), 2.36 (s, 3H, CH2–S–C3), 2.13–2.06 (m, 2H, CH–C2–CH2–), 1.95–1.84 (m, 2H, CH–CH2–C2–). 13C NMR (100 MHz, CDCl3) δ, ppm: 195.65 (–S–O–CH3), 112.53 (–N), 30.76 (–S–C(O)–H3), 29.55 (CH2–H2–CH2), 27.22 (H2–SAc), 26.84 (CH–H2–), 22.30 (–H–CH2–). MS (EI+) m/z (%): 182.05 (5.53), 43.02 (100). EA: calculated: [C] 52.73% [H] 5.53% [N] 15.37% [O] 8.78% [S] 17.59% found: [C] 52.53% [H] 5.75% [N] 15.31% [O] 9.05 [S] 17.53%.–CH2–), 2.61–2.67 (m, 2H, CH2–C2–SH), 2.23–2.17 (m, 2H, CH–C2–CH2–), 1.98–1.90 (m, 2H, CH–CH2–C2–), 1.43 (t, 3J = 8.1 Hz, 1H, –CH2–S). 13C NMR (100 MHz, CDCl3) δ, ppm: 195.60 (–S–O–CH3), 112.54 (–N), 29.47 (CH2–H2–CH2), 30.39 (H2–SH), 23.39 (CH–CH2–), 22.46 (–CH–H2–). HRMS (ESI): calc. C6H9N2S m/z: 141.0481 [M + H]; found 141.0481 [M + H].:1). 1H NMR (400 MHz, CDCl3) δ, ppm: 4.33 (br s, 2H, –NH2), 2.94 (t, 3J = 5.6 Hz, 2H, –CH2–C2–S), 2.35 (t, 3J = 6.4 Hz, 2H, –C2–CH2–CH2–), 2.03–1.96 (m, 2H, –CH2–C2–CH2). 13C NMR (100 MHz, CDCl3) δ, ppm: 154.19 (C(–NH2)–S), 120.13 (–N), 72.94 ((NC)–C–S), 28.17 (S–H2–CH2–CH2–), 24.37 (S–CH2–CH2–H2–), 22.47 (–CH2–H2–CH2–). HRMS (ESI) calc. for C6H9N2S [M + H] 141.0481, found 141.0481. EA: calculated: [C] 51.40% [H] 5.74% [N] 19.98% found: [C] 51.44% [H] 5.83% [N] 19.73%.:1) to give a pale yellowish solid (9.0 g). The solid was dissolved in ethyl acetate and precipitated by slow addition n-heptane to obtain 9 (8.2 g, 31%) as colourless solid. RF = 0.5 (heptane/ethyl acetate 1:1). 1H NMR (400 MHz, CDCl3) δ, ppm: 5.67 (d, 3J = 16 Hz, 1H, CHCH–), 5.58 (d, 3J = 11 Hz, 1H, CHCH–), 5.44 (dd, 3J = 10.2 Hz, 3J = 16.8 Hz, 1H, CH2C–), 2.57–2.39 (m, 4H, CH2–C2–CN), 2.24–2.17 (m, 2H, CH–CH2–CN), 2.00–1.92 (m, 2H, CH–CH2–CN). 13C NMR (100 MHz, CDCl3) δ, ppm: 132.76 (CH2H–), 122.17 (H2CH–), 117.98 (Cquart–N), 117.85 (CH2–N), 45.81 (Cquart), 34.22 (H2–CH2CN), 13.66 (CH2–H2–CN). MS (EI) m/z (%): 55.05 (49.53), 80.05 (34.74), 92.05 (70.69), 119.06 (100). EA: calculated: [C] 69.34% [H] 6.40% [N] 24.26% found: [C] 69.21% [H] 6.33% [N] 24.16%.:1) to give (0.4 g, 58%) of 12. RF = 0.2 (heptane/ethyl acetate 1:1). 1H NMR (400 MHz, acetone-d6) δ, ppm: 8.68 (br s, 4H, –CS–N2), 5.71 (dd, 3J = 10 Hz, 3J = 17 Hz, 1H, CH2CH–), 5.42 (d, 3J = 17 Hz, 1H, CHCH–), 5.35 (d, 3J = 10 Hz, 1H, CHCH–), 2.77–2.63 (m, 4H, CH2–C2–C(S)NH2), 2.28–2.11 (m, 4H, C2–CH2–CN). 13C NMR (100 MHz, acetone-d6) δ, ppm: 208.12 (–(S)NH2), 136.79 (CH2H–), 120.21 (H2CH–), 117.37 (Cquart–N), 45.60 (Cquart), 40.14 (H2–H2C(S)NH2), 37.31 (CH2–H2–C(S)NH2); MS (ESI): calc. for C10H16N3S2 [M + H] 242.0780; found 242.0779.2–C2–SH), 2.68 (t, 3J = 7.2 Hz, 2H, –S–C2–CH2–), 2.51 (t, 2H, –CH2–C2–CN, J = 6.8 Hz), 1.94 (dt, 4J = 7.44 Hz, 2H, –CH2–C2–CH2–CN), 1.71 (t, 3J = 8.1 Hz, 1H, –CH2–S). 13C NMR (100 MHz, CDCl3) δ, ppm: 119.27 (CH2–N), 36.20 (HS–CH2–H2–S–), 30.66 (–S–H2–CH2–), 25.36 (–CH2–H2–CH2–), 24.77 (HS–H2–CH2–), 16.14 (–CH2–H2–CN). MS (EI+) m/z (%): 114.03 (94.2), 161.03 (60.5). EA: calculated: [C] 44.68% [H] 6.87% [N] 8.68% [S] 39.76 found: [C] 44.51% [H] 7.12% [N] 8.58% [S] 39.78.–CH2–), 2.79–2.69 (m, 4H, –S–(C2)2–SH), 2.64 (t, 3J = 7.1 Hz, 2H, CH2–C2–S–), 2.21–2.15 (m, 2H, CH–C2–CH2), 1.96–1.88 (m, 2H, CH2–C2–CH2), 1.72 (t, 3J = 8.0 Hz, 1H, CH2–S–). 13C NMR (100 MHz, CDCl3) δ, ppm: 112.47 ((N)2–CH–), 36.11 (–H2–CH2–SH), 30.62 (–H2–CH2–S–), 29.64 (CH–H2–CH2), 26.11 (CH2–H2–CH2), 24.72 (CH2–H2–SH), 22.48 ((CN)2–H–). MS (ESI) m/z (%): 167 (100), 359.24 (22.5), 423 (23.8). EA: calculated: [C] 47.97% [H] 6.04% [N] 13.98% [S] 32.01%/found: [C] 47.90% [H] 6.24% [N] 13.74% [S] 31.86%.:1) to give (0.09 g, 34%) 15 as yellowish liquid. 1H NMR (400 MHz, CDCl3) δ, ppm: 2.83–2.72 (m, 4H, HS–C2–C2–S), 2.67 (t, 2H, –S–C2–CH2–Cquart, 3J = 8 Hz), 2.57 (t, 3J = 8 Hz, 4H, –CH2–C2–CN), 2.07 (t, 3J = 8 Hz, 4H, –C2–CH2–CN), 1.96 (t, 3J = 8 Hz, 2H, –C2–Cquart), 1.73 (t, 1H, –S). 13C NMR (100 MHz, CDCl3) δ, ppm: 119.96 (Cquart–N), 117.89 (–CH2–N), 40.62 (quart), 36.65 (–H2–SH), 35.78 (S–CH2–H2–Cquart–), 31.63 (H2–CH2–CN), 26.79 (S–H2–CH2–Cquart–), 24.89 (HS–CH2–H2–S), 13.48 (H2–CN). MS (EI+) m/z (%): 61.01 (100), 69.05 (15.55), 207.08 (17.18), 267.08 (13.02). EA: calculated: [C] 53.90% [H] 6.41% [N] 15.71% [S] 23.98 found: [C] 53.78% [H] 6.60% [N] 15.43% [S] 23.82%.Compounds 16 & 17
To a solution of allyl malononitrile (53 g, 0.5 mol) dissolved in freshly distilled THF (500 ml), 1,2-ethanedithiol (235 ml, 2.5 mol) and DMPA (2.5 g, 0.002 mol) was added under argon atmosphere. The reaction mixture was irradiated for 5 min. All volatiles were removed at 80 °C in vacuo (10 mbar). The residue was purified by SiO2 column chromatography (heptane/ethyl acetate → methanol → DMF) to give 17 (2 g, 2%) as colourless crystals and 16 (40 g, 40%) as pale brownish, viscous liquid.–CH2–), 2.79–2.69 (m, 4H, –S–(C2)2–SH), 2.64 (t, 3J = 7.1 Hz, 2H, CH2–C2–S–), 2.21–2.15 (m, 2H, CH–C2–CH2), 1.96–1.88 (m, 2H, CH2–C2–CH2), 1.72 (t, 3J = 8.0 Hz, 1H, CH2–S–). 13C NMR (100 MHz, CDCl3) δ, ppm: 112.47 ((N)2–CH–), 36.11 (–H2–CH2–SH), 30.62 (–H2–CH2–S–), 29.64 (CH–H2–CH2), 26.11 (CH2–H2–CH2), 24.72 (CH2–H2–SH), 22.48 ((CN)2–H–).2), 6.37 (br s, –N2), 3.20–3.30 (m, 2H, –CH2–C2–S–C), 2.74–2.79 (m, 2H, CH2–S–C2–CH2), 2.61–2.68 (m, 2H, CH2–C2–CH2–S), 1.69–1.81 (m, 2H, CH2–C2–CH2), 2.27–2.40 (m, 2H, C–C2–CH2). 13C NMR (100 MHz, N,N-dimethyformamide-d7) δ, ppm: 152.17 (C(NH2)–S), 122.13 (C–N), 80.72 (C(CN)–), 31.97 (CH2–H2–S–C), 31.21 (H2–CH2–S–C), 30.53 (CH2–CH2–H2–S), 28.55 (CH2–H2–CH2–S), 27.16 (H2–CH2–CH2–S). HRMS (ESI): calc. for C16H25N4S4 [M + H] 401.0957; found: 401.0957.Results and discussion
Allyl cyanide is the simplest aliphatic non-conjugated vinyl nitrile. The thiol–ene addition of A to allyl cyanide initiated by 1 mol% DMPA gave S-3-cyanopropyl thioacetate 1 in 73% isolated yield within 5 min (Scheme 1). Compound 1 is known in the literature and was prepared starting from potassium salt of A and 4-bromobutyronitrile. Unfortunately, no reaction yield is given for this reaction.34,35 Cleavage of the acetyl moiety of 1 under acidic conditions (20 mol% trimethylsilyl chloride (TMSCl) in methanol) proceeded at low reaction rate and gave 4-mercaptobutanenitrile 2 in moderate yields (43%). When a stoichiometric amount of TMSCl was used, 2-iminothiolane hydrochloride 3 formed in 60% yield. Compound 2 is known and was prepared starting from 4-isothioureidobutyronitrile hydrochloride but the reaction yield was very low (15%).36 Compound 3 is also known in the literature and was prepared by nucleophilic substitution starting from 4-halogenobutanenitrile and A or thiourea, respectively.37–39 It is widely used as thiolation reagent in biochemistry and is referred to as ‘Traut's reagent’.40–43 | ||
| Scheme 1 Thiol–ene addition of A to allyl cyanide followed by the cleavage of the acetyl group to give 2 and 3. | ||
Allyl malononitrile 5 was synthesized according to the literature starting from malononitrile and allyl bromide in one step (Scheme 2).44 The thiol–ene addition of a slight excess of A to 5 in presence of DMPA gave S-(4,4-dicyanobutyl)thioacetate 6 in 74% yield within 5 min (Scheme 2). When a large excess of A was used, addition to the nitrile groups was observed. Acetate cleavage under basic conditions in presence of butyl amine resulted in quantitative cyclization of 6 to 6-amino-3,4-dihydro-2H-thiopyran-5-carbonitrile 8. The quantitative conversion of 6 to 8 is supported by the 1H NMR spectra where the characteristic signals of 6 disappeared and new signals appeared which can be easily assigned to 8 (Fig. 1). Single crystals of 8 suitable for X-ray analysis were grown from ethyl acetate/heptane. The proposed six-membered ring was confirmed by X-ray structure (Fig. 2). The transesterification reaction with methanol under acidic conditions allowed formation of 4,4-dicyanobutyl thiol 7 but the reaction did not proceed to completion. Attempts to purify this compound by vacuum distillation were only partially successful. The compound still contained about 15% impurities (Fig. 1d). Further attempts to increase the purity of 7 by column chromatography purification on silica gel were unsuccessful. The intramolecular cyclization of 7 occurred quantitatively with formation of 8 (Fig. 1). The synthesis of the analog five-membered cycle starting from malononitrile and ethylene sulfide was already described in the literature, but an over-stoichiometric amount of a strong base such as sodium hydride had to be used and the reaction yield was only moderate (57%).45
 | ||
| Scheme 2 Synthetic route to 4,4-dicyanobutyl thiol 7. | ||
 | ||
| Fig. 1 1H NMR spectra of: (a) 6, (b) of the crude reaction mixture obtained by reacting 6 with butyl amine, (c) of 8, and (d) of 7. | ||
 | ||
| Fig. 2 ORTEP plot of 8, view perpendicular to the cycle (left) and the molecular packing down the crystallographic b axis showing the stacking of the cycles and the hydrogen bonding network (right). | ||
3-Vinylpentane-1,3,5-trinitrile 9 was synthesized according to literature starting from allyl cyanide (Scheme 3).46 The vinyl group of 9 is sterically hindered due to the bulky –(CH2)2CN moieties in alpha position to the vinyl group. This is nicely illustrated by the 1H NMR spectrum of 9 where the two protons of the –CH2–CH2–CN group next to the quaternary carbon are not equivalent, as one would expect (see ESI†). This indicates a lack of free rotation of the –(CH2)2CN moiety. The thiol–ene addition of A to 9 initiated by DMPA did not occur even after 60 min UV irradiation. Instead, instability of the nitriles was observed. When AIBN as thermal radical initiator and excess of A was used, the addition of A to the nitrile group was observed. NMR analysis of the crude product mixture showed the formation of 11 which hydrolysed to 4-cyano-4-vinylheptane bis(thioamide) 12 during work-up (60% yield). Formation of 3-(2-thioacetic)ethylpentane-1,3,5-carbonitrile 10 was not observed. The conversion of aliphatic nitriles to thioamides is known, however this occurs either under acidic or basic catalysis.30,31 At 70 °C the acidity of A was sufficient to catalyse its addition to the nitrile of 9.
 | ||
| Scheme 3 Synthesis of 9 by cyanoethylation of allyl cyanide and its reaction with AcSH. | ||
As an alternative to A in the transformation of vinyl nitriles to mercaptonitriles, 1,2-ethanedithiol B was tested (Scheme 4). The advantage of B is that it allows formation of mercaptonitriles directly via thiol–ene reaction of vinyl nitriles, even though an excess of B has to be used and the spacer between the thiol and the nitrile group is prolonged. Intramolecular cyclization reactions of the formed mercaptonitriles are less favored as five or six-membered rings cannot form.
 | ||
| Scheme 4 Thiol–ene addition of 1,2-ethanedithiol to vinyl nitriles. | ||
Addition of B to allyl cyanide gave 4-((2-mercaptoethyl)thio)butanenitrile 13, which did not show any susceptibility to cyclization. The purification of 13 was done by distillation and the compound could be easily prepared on 30 g scale. The reaction of 5 with B gave 2-(3-((2-mercaptoethyl)thio)propyl)malononitrile 14. The isolation of 14 by distillation was possible, but challenging given 14's high boiling point. Again, silica gel during chromatography purification catalysed the intramolecular cyclization with formation of cyclic dimer 17 in small amounts. The structure of 17 was confirmed by X-ray diffraction analysis on a single crystal (Fig. 3). The main product after chromatography was oligomer 16 which was obtained via silica gel extraction with DMF and methanol as can be seen in the 1H NMR spectrum in the ESI.† No further characterizations were conducted on 16 due to its poor solubility.
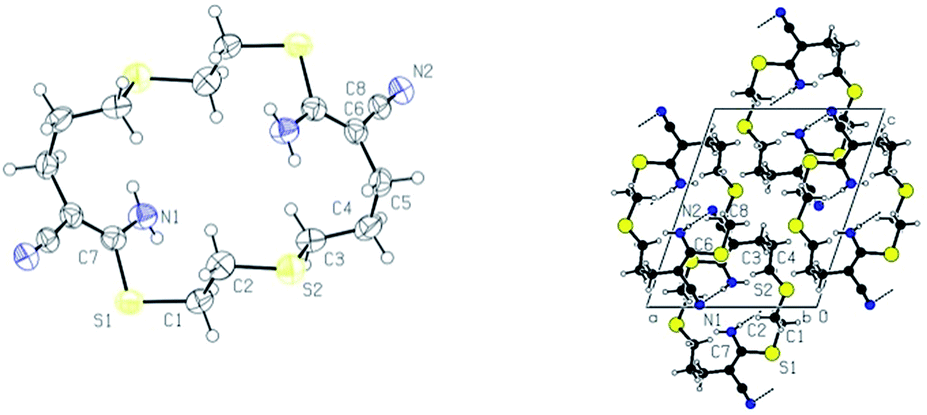 | ||
| Fig. 3 ORTEP plot of 17, view perpendicular to the cycle (left) and the molecular packing down the crystallographic a axis showing the hydrogen bonding network (right). | ||
In contrast to A, selective thiol–ene addition of B to the vinyl group of 9 was achieved by using B in excess, an over-stoichiometric amount of photoinitiator, and prolonged reaction times. The difference in the reactivity of A and B can be explained by the acidity of the two reagents used.47 Due to the presence of the electron-withdrawing group, A is more acidic as compared to 1,2-ethanedithiol and therefore chemically more reactive towards nitrile. NMR analysis of the reaction mixture showed product conversion of approximately 50%. The nitrile groups were unaffected during reaction and chromatography purification process. The mercaptonitriles 13–15 did not crystallize. Traces of Markovnikov addition product were detected in all cases.
Thiols 13 and 14 were identified as being promising candidates for the post polymerization modification of polyvinylsiloxanes via thiol–ene addition. 13 and 14 can be prepared easily on a 20 g scale and can be purified by distillation. Although the high boiling points of these thiols may be considered disadvantageous since high vacuum and high temperatures have to be used for their distillation, they have the advantage that they are less volatile and therefore do not have the typical pungent odor characteristic for thiols. The thiols show a dielectric permittivity at high frequencies of ε′ = 15.4 and ε′ = 17 for 13 and 14, respectively. Preliminary results show that 13 and 14 can be successfully used in the selective thiol–ene addition to polyvinylsiloxane and no side reactions were observed. Further work in our lab is conducted in this direction.
Conclusions
In conclusion, the thiol–ene addition of A and B to allyl cyanide, allyl malononitrile, and 3-vinylpentane-1,3,5-carbonitrile was investigated. All vinyl nitriles reacted with A and B as expected except 3-vinylpentane-1,3,5-carbonitrile. This compound with its sterically hindered vinyl group did not give the thiol–ene addition product A but the addition product to the nitriles. The cleavage of the acetyl groups to generate the respective mercaptonitriles must be conducted under mild acidic conditions due to the high tendency of the generated thiols to undergo addition to the nitriles under acidic and basic conditions. Purification of the mercaptonitriles by column chromatography over silica gel should be avoided. Thiol–ene addition of B to non-conjugated vinyl nitrile compounds proved to be a fast and efficient alternative for A in the synthesis of mercaptonitriles in one step and mild reaction conditions.Acknowledgements
We gratefully acknowledge Swiss Federal Laboratories for Materials Science and Technology (Empa, Dübendorf) for financial support.Notes and references
- V. N. Ipatieff and B. S. Friedman, J. Am. Chem. Soc., 1939, 61, 71 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed.
- L. Gu, Q. Xue, S. Peng, G. Wang, J. Han and X. Wu, Polymer, 2016, 7, 625 CAS.
- C. Ligeour, L. Dupin, A. Marra, G. Vergoten, A. Meyer, Y. Dondoni, A. Souteyrand, E. Vasseur, J.-J. Chevolot and F. Morvan, Eur. J. Org. Chem., 2014, 7621 CrossRef CAS.
- Z. Liu, T. Liu, Q. Lin, C. Bao and L. Zhu, Angew. Chem., Int. Ed., 2015, 54, 174 CrossRef CAS PubMed.
- Y. Suzuki, T. Higashihara, S. Ando and M. Ueda, Macromolecules, 2012, 45, 3402 CrossRef CAS.
- N. K. Singha, M. I. Gibson, B. P. Koiry, M. Danial and H.-A. Klok, Biomacromolecules, 2011, 12, 2908 CrossRef CAS PubMed.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14 CrossRef CAS.
- Y. Chen, G. Chen and M. H. Stenzel, Macromolecules, 2010, 43, 8109 CrossRef CAS.
- J. Z. Du, X. J. Du, C. Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560 CrossRef CAS PubMed.
- M. Podgorski, E. Becka, M. Claudino, A. Flores, P. K. Shah, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2015, 31, 1255 CrossRef CAS PubMed.
- E. Klemm and S. Sensfuss, Makromol. Chem., 1991, 64, 159 CrossRef.
- A. S. Quick, J. Fischer, B. Richter, T. Pauloehrl, V. Trouillet, M. Wegener and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 335 CrossRef CAS PubMed.
- C. S. Marvel and A. Kotch, J. Am. Chem. Soc., 1951, 73, 1100 CrossRef CAS.
- S. J. Duenki, Y. S. Ko, F. A. Nueesch and D. M. Opris, Adv. Funct. Mater., 2015, 25, 2467 CrossRef CAS.
- S. J. Dünki, M. Tress, F. Kremer, S. Y. Ko, F. A. Nüesch, C.-D. Varganici, C. Racles and D. M. Opris, RSC Adv., 2015, 5, 50054 RSC.
- C. Decker and T. Nguyen Thi Viet, Polymer, 2000, 41, 3905 CrossRef CAS.
- A. Gress, A. Vo and H. Schlaad, Macromolecules, 2007, 40, 7928 CrossRef CAS.
- J. C. Persson, K. Josefsson and P. Jannasch, Polymer, 2006, 47, 991 CrossRef CAS.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854 RSC.
- D. M. Opris, E. Perju and S. J. Duenki, J. Polym. Sci., Part A: Polym. Chem., 2016, 56, 2940 Search PubMed.
- M. Le Neindre, B. Magny and R. Nicolaÿ, Polym. Chem., 2013, 4, 5577 RSC.
- J. Polster and P. Schieberle, J. Agric. Food Chem., 2015, 63, 1419 CrossRef CAS PubMed.
- E. Schjanberg, Ber. Dtsch. Chem. Ges., 1941, 74, 1751 CrossRef.
- R. Brown, W. E. Jones and A. R. Pinder, J. Chem. Soc., 1951, 2123 RSC.
- J. Gorges and U. Kazmaier, Eur. J. Org. Chem., 2015, 8011 CrossRef CAS.
- J. W. Lynn, R. L. Roberts and J. R. Kilsheimer, J. Org. Chem., 1961, 26, 4300 CrossRef CAS.
- A. Kjaer and J. Conti, Acta Chem. Scand., 1954, 8, 295 CrossRef CAS.
- M. Yunhai and X. Jiaxi, Synthesis, 2012, 2225 Search PubMed.
- J. Y. Gauthier and H. Lebel, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 95, 325 CrossRef.
- K. A. Mahammed, V. P. Jayashankara, N. Rai, K. Raju, K. Mohana and P. N. Arunachalam, Synlett, 2009, 2338 CAS.
- R. Jue, J. M. Lambert, L. R. Pierce and R. R. Traut, Biochemistry, 1978, 17, 5399 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
- N. Park, K. Park, M. Jang and S. Lee, J. Org. Chem., 2011, 76, 4371–4378 CrossRef CAS PubMed.
- M. Los, American Cyanamid Co., Eur. Pat. Appl. EP 133310, 1985.
- R. R. Traut, A. Bollen, T. T. Sun, J. W. B. Hershey, J. Sundberg and L. R. Pierce, Biochemistry, 1973, 12, 3266 CrossRef CAS PubMed.
- M. Wallace, A. J. Allentoff, J. Brailsford, S. Gong, S. Bonacorsi and F. Rinaldi, J. Labelled Compd. Radiopharm., 2015, 58, 429 CrossRef CAS PubMed.
- S. F. Carroll and D. A. Goff, WO 90/06774, 1990.
- T. P. King, Y. Li and L. Kochoumian, Biochemistry, 1978, 17, 1499 CrossRef CAS PubMed.
- T. P. King, Y. Li and L. Kochoumian, Biochemistry, 1986, 25, 5774 CrossRef CAS PubMed.
- A. Braden, M. Roner, J. Ganter and K. Nelson, J. Nanosci. Nanotechnol., 2007, 7, 925 CrossRef CAS PubMed.
- T. Tada, K. Mano, E. Yoshide, N. Tanaka and S. Kunugi, Bull. Chem. Soc. Jpn., 2002, 75, 2247 CrossRef CAS.
- T. T. Ngo, Appl. Biochem. Biotechnol., 1986, 13, 213 CrossRef CAS.
- M. K. Ghorai, R. Talukdar and D. P. Tiwari, Org. Lett., 2014, 16, 2204 CrossRef CAS PubMed.
- K. Yamagata, T. Yukihiko, M. Yamazaki, T. Matsuda and K. Noda, Chem. Pharm. Bull., 1982, 30, 4396 CrossRef CAS.
- H. A. Bruson and T. W. Riener, J. Am. Chem. Soc., 1943, 65, 18 CrossRef CAS.
- B. Dmuchovsky, F. B. Zienty and W. A. Vredenbur, J. Org. Chem., 1966, 31, 865 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization of all new compounds, X-ray crystallographic data for 8, 9, and 17, and the dielectric spectra of 13 and 14 are given. CCDC 1492250–1492252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra21948a |
| This journal is © The Royal Society of Chemistry 2016 |