
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Attachment of antimicrobial peptides to reverse osmosis membranes by Cu(I)-catalyzed 1,3-dipolar alkyne–azide cycloaddition
Elias J.
Bodner
ae,
Nitzan Shtreimer
Kandiyote
a,
Marina-Yamit
Lutskiy
a,
H. Bauke
Albada
 bc,
Nils
Metzler-Nolte
b,
Wolfgang
Uhl
de,
Roni
Kasher
*a and
Christopher J.
Arnusch
*a
bc,
Nils
Metzler-Nolte
b,
Wolfgang
Uhl
de,
Roni
Kasher
*a and
Christopher J.
Arnusch
*a
aDepartment of Desalination and Water Treatment, Zuckerberg Institute for Water Research, The Jacob Blaustein Institutes for Desert Research, Ben-Gurion University of the Negev, Sede-Boqer Campus 84990, Israel. E-mail: arnusch@bgu.ac.il; kasher@bgu.ac.il; Tel: +972-8-656-3532 Tel: +972-8-656-3531
bInorganic Chemistry I, Bioinorganic Chemistry, Faculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstrasse 150, 44801 Bochum, Germany
cLaboratory for Organic Chemistry, Wageningen University, The Netherlands
dNorwegian Institute for Water Research (NIVA), Gaustadalléen 21, 0349 Oslo, Norway
eChair of Water Supply Engineering, Technische Universität Dresden, 01062 Dresden, Germany
First published on 21st September 2016
Abstract
Biofilms are detrimental to many industrial systems that include reverse osmosis (RO) membranes. Accordingly, the development of surfaces with inherently bactericidal properties has attracted much research attention. Antimicrobial peptides (AMPs) have been shown to be potent antimicrobial and anti-biofilm agents. In the current study, we developed an efficient synthetic procedure for AMP immobilization on RO membranes which is based on the copper(I) mediated Huisgen 1,3-dipolar cycloaddition reaction (“click chemistry”). Optimization of the reaction temperature, time, peptide and catalyst concentration resulted in efficient coupling of peptides to the membrane surface. The reaction conditions did not affect membrane salt rejection, and resulted in only a slight reduction (14%) in pure water flux at the highest temperature tested (80 °C). Short AMPs that consisted of Arg–Trp repeats were attached onto a virgin RO membrane surface, and an RO membrane surface coated with a copolymer of methacrylic acid and poly(ethylene glycol)methacrylate. In a bacterial contact killing assay, the resulting peptide-modified membrane surfaces showed increased antimicrobial activity especially on the virgin membrane as compared to unmodified membranes. This study provides a basis for further research into the attachment of a wide variety of antimicrobials or other entities to surfaces.
1. Introduction
In order to supply drinking water to growing populations, even in areas of the world where fresh water is scarce, desalination technologies have been attracting considerable attention as effective methods for the production of potable water.1,2 Among them, reverse osmosis (RO) membrane technology today accounts for the major share of potable water production from seawater and brackish water.3 However, biofouling, i.e. the formation of biofilms on the membrane surfaces, is a major problem as it brings about serious impairments of the overall membrane performance: increased trans membrane pressure and thus energy consumption, decreased flux, increased salt passage, shortened membrane lifespan and consequently in higher operational costs than for non-fouled membranes.4–6 In order to mitigate these drawbacks, the feed water can be pretreated in order to remove bacteria and dissolved substances, which can serve as nutrients to bacteria attached to the membranes. But these are only partially effective as they cannot completely eliminate the organic matter responsible for fouling and the concentrations of microorganisms and nutrients that remain in the treated feed water are high enough to promote bacterial growth on the membrane surface.7The problem of biofilm formation is ubiquitous – from industrial water treatment settings to biomedical applications to natural environments – due to the ability of the microorganism to colonize almost any surface where nutrients tend to concentrate and accumulate.4 It begins with deposition of bacteria on the membrane surface and adhesion, followed by multiplication and micro-colony formation, and is accompanied by the excretion of extracellular polymeric substances (EPS). Once embedded in the EPS, the microbial cells are practically irreversibly attached to the surface and difficult to remove.8–10
As the attachment of bacteria to the surface is the initial step of biofouling, an intriguing strategy for combatting biofilm formation focuses on the physical and functional characteristics of the surface. Thin film composite (TFC) polyamide membranes, widely used in RO applications, consist of a dense, semipermeable polyamide barrier layer (50–200 nm) supported on a polysulfone layer (∼40 μm) on a fibrous polyester web (∼120 μm) that provides mechanical stability. The major transport properties of these membranes, determined by the polyamide barrier layer, include the almost complete retention of salts (salt rejection above 99%) and a substantial flux of water.3–11 Biofilm inhibition strategies for such membranes have included surface modifications and coatings, since membrane surface characteristics significantly affect foulant deposition and fouling layer formation. For example, Rahaman et al. observed a reduction in biofouling on TFC polyamide RO membranes modified with biocidal silver nanoparticles and antifouling polymer brushes.12 The surface modifications resulted in a significant reduction of irreversible bacterial cell adhesion. Nikkola et al. studied polyvinyl alcohol coatings modified with cationic polyhexamethylene guanidine hydrochloride polymer for enhanced anti-adhesion and anti-biofouling membranes.13 Zodrow et al. used a controlled-release platform, in which antibacterial compounds (cinnamaldehyde, kanamycin) were encapsulated in biodegradable poly(lactic-co-glycolic acid) particles and bound to TFC polyamide RO membranes, to achieve significant reductions in biofilm formation.14
Beyond that, it is even more intriguing to modify membrane surfaces such that they exhibit antimicrobial properties by inhibiting bacterial proliferation. A strategy that prevents viable bacteria from settling and multiplying on the surface may halt the biofilm formation process in its early stages. This approach was followed, as the objective herein was to optimize the covalent binding of antimicrobial peptides (AMPs) to membrane surfaces. Such a tool may facilitate the development of membranes with inherent antimicrobial properties. AMPs have been shown to have anti-biofilm properties.15 The covalent immobilization of the AMP also ensures that the active coating will not eventually leach from the membrane surface.
Examples of antimicrobial peptides covalently tethered to different surfaces were reviewed by Costa et al., who also discussed the various methods for covalently immobilizing AMPs onto biomaterial surfaces, including important parameters such as spacer length, peptide concentration and orientation.16 These studies showed that the immobilization of AMPs on surfaces may prevent biofilm formation by reducing microbial viability after it comes in contact with the AMP-containing surface. The attachment of AMPs on the surfaces of RO membranes has been previously studied for effective antimicrobial surfaces17 and suggested as a method of biofilm inhibition.18
The aim of this study was to optimize AMP attachment to TFC polyamide RO membranes via a copper(I) mediated Huisgen 1,3-dipolar cycloaddition reaction between an azide and an alkyne (CuAAC, “click chemistry”). Widely adopted as an efficient conjugation method for a broad range of materials and compounds,19–21 click chemistry has previously been used for membrane materials such as polysulfone, polypropylene, and polyamide.22–24 The advantages of this reaction include high yield, broad scope, stereospecificity, and that it can be conducted in aqueous solvents under mild conditions.25–29 In comparison to bonding the peptide to the membrane via an amide linkage, the resulting 1,4-disubstituted 1,2,3-triazoles have been shown to be similar to the amide moiety in terms of size, planarity, H-bonding capabilities, and dipole moment, while being more protease-resistant.30 Previously, click reactions were exploited to modify polyamide membrane surfaces through the attachment of polyzwitterions onto polyamide, which enhanced membrane fouling resistance. The key steps included synthesis of an alkyne-polyzwitterion via reversible addition–fragmentation chain transfer polymerization, functionalization of polyamide with azide functional groups by bromination followed by the SN2 nucleophilic substitution of Br with azide, and finally, the attachment of alkyne-polyzwitterion onto azide-functionalized polyamide by an azide–alkyne cycloaddition. Improved antifouling of the polyamide membrane was observed, a finding attributable to the effects of reduced specific binding, steric repulsions and strong hydrophilic repulsion.31
In the present study, we functionalized the polyamide surface of commercial RO membranes with a short azide linker. Alkyne functionalized AMPs were synthesized using the silyl-based alkyne modifying-linker, which conveniently gives C-terminal acetylene-derivatized peptides upon cleavage from the resin. The mode of surface attachment of the alkyne functionalized peptides to the azide functionalized RO membranes was optimized by varying reaction conditions, including time, temperature, and the concentrations of reagents and catalyst, taking into consideration the effect of the reaction conditions on membrane performance in terms of salt rejection and pure water permeability. This optimization was performed with short AMPs on RO membrane surfaces directly and on a surface-grafted layer consisting of a copolymer of methacrylic acid and poly(ethylene glycol)methacrylate. The functionalized surfaces were then tested for antimicrobial activity via a bacterial contact killing assay. The peptide modified non-grafted surfaces showed antibacterial activity in comparison to non-modified membranes. The methods and findings presented herein provide a basis for future studies to examine the attachment of a wide variety of antimicrobials and other entities to surfaces.
2. Experimental section
2.1. General
Flat-sheet extra low energy polyamide RO membranes (XLE membrane) were provided by DOW FILMTEC Membranes (Midland, MI). Poly(ethylene glycol)methacrylate (PEGMA, product # 409537), methacrylic acid (MA, 99%), sodium hydroxide (NaOH) and cupric sulfate (CuSO4, 99%) were purchased from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Potassium metabisulfite (K2S2O5, 97%) and potassium persulfate (K2S2O8, 99%) were obtained from Acros Organics (Geel, Belgium). Ethyl(dimethylaminopropyl)carbodiimide (EDC, 99.9%), N-hydroxysuccinimide (NHS, 99.9%), and sodium ascorbate (99.6%) were purchased from Chem-Impex International, Inc. (Wood Dale, IL). Isopropyl alcohol and ethanol were purchased from Merck Millipore (Darmstadt, Germany). Phosphate buffered saline (PBS) was prepared in the laboratory with sodium chloride (NaCl), potassium chloride (KCl), disodium phosphate (Na2HPO4) and potassium phosphate (KH2PO4) purchased from Bio-Lab Ltd. (Jerusalem, Israel). Lysogeny broth (LB) was prepared with Bacto Tryptone and Bacto Yeast Extract obtained from Becton, Dickinson and Company (New Jersey, United States). Sodium chloride (NaCl) and hydrochloric acid (HCl) were purchased from Bio-Lab Ltd. (Jerusalem, Israel). Bacto Agar was obtained from Becton, Dickinson and Company (New Jersey, United States).For contact angle measurements using the sessile drop method, the OCA-20 contact angle analyzer (DataPhysics, Filterstadt, Germany) was used. Prior to that, membrane samples were dried under vacuum for at least 3 h at room temperature. For each membrane sample at least five measurements were done, using drops of 0.5 μL distilled water, and the average and standard deviation were calculated.
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) was carried out for at least five times per dried membrane sample using a Vertex 70 spectrometer (Bruker Optics, Ettlingen). The data were evaluated using the spectral analysis software (OPUS 6.5), averages were compiled and the ratios and standard deviation of the signal intensities at different wavelengths were calculated.
X-ray photoelectron spectroscopy (XPS) was carried out on dried membranes using an ESCALAB 250 apparatus (Thermo Fisher Scientific).
2.2. Synthesis of peptides
Although it was based on a procedure described previously,32 the synthesis of peptide 1 in this study employed 4-pentynoic acid as the acylating agent for the lysine side-chain amino group. Peptides 2 and 3 were synthesized using standard, published procedures with the second generation silyl-based alkyne modifying (SAM2)-linker.33 In short, the SAM2-linker was immobilized on a polystyrene-aminomethyl resin using a reductive amination reaction. Then, all reactive amino-groups were permanently capped with acetyl-groups, and the SAM2-protecting trityl-group was removed using diluted trifluoroacetic acid (TFA) in CH2Cl2. The exposed amino-group was the anchor-point on which the Arg–Trp based synthetic AMP precursor was built.34 Since the two peptides share the C-terminal pentapeptide sequence, this peptide was assembled on the entire batch. After the H–Trp(Boc)–Arg(Pbf)–Trp(Boc)–Arg(Pbf)–Trp(Boc) sequence was assembled on the SAM2-polystyrene resin, the batch was divided into two portions: (1) for the coupling of ferrocene carboxylic acid (i.e., FcC(O)OH), and (2) for the coupling of Fmoc–Arg(Pbf)–OH. For the latter of the two portions, synthesis of the resin-bound peptide was completed by Fmoc-deprotection. Lastly, the FcC(O)–Trp–Arg–Trp–Arg–Trp–N(H)CH2CCH peptide was obtained by cleaving the peptide in the presence of triisopropylsilane (TIS) and phenol; for the H–(Arg–Trp)3–N(H)CH2CCH peptide, the standard TFA/TIS/water mixture was applicable.2.3. Analysis of peptides
Analytical high performance liquid chromatography (HPLC) was performed on an automated HPLC system using a C18-AQ reverse phase (RP) column (250 × 4.6 mm) at a flow-rate of 1 mL min−1: 0–5 min 100% buffer A; 5–25 min linear gradient (0–100%) of buffer B; 25–30 min 100% buffer B; 30–35 min linear gradient (100–0%) of buffer B; 35–40 min 100% buffer A (buffer A: H2O/acetonitrile/TFA, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:0.1, v/v/v; buffer B: acetonitrile/H2O/TFA, 95:5:0.1, v/v/v). Purification of the peptides was performed on an HPLC machine equipped with photodiode array detector that was coupled to an RP-18e reversed phase column (250 × 25 mm), using a similar gradient as for the analytical HPLC but with a flow rate of 20 mL min−1. The cleaved peptides were >90% pure (as inferred from HPLC), and were obtained as pure compounds after preparative HPLC. Analysis of peptide 1 (H–(Arg–Trp)3–Lys(C(O)CH2CH2CCH)–NH2): RP-HPLC (C18): tR = 17.50 min; ESI-MS: m/z 627.18 (calc. 627.26 for [M + H]2+). Peptide 2 (H–(Arg–Trp)3–N(H)CH2CCH): RP-HPLC (C18): tR = 17.6 min; ESI-MS: m/z 1082.20 (calc. 1082.59 for [M + H]+). Peptide 3 (FcC(O)–Trp–(Arg–Trp)2–N(H)CH2CCH): RP-HPLC (C18): tR = 20.3 min; ESI-MS: m/z 1138.08 (calc. 1138.48 for [M + H]+).
:5:0.1, v/v/v; buffer B: acetonitrile/H2O/TFA, 95:5:0.1, v/v/v). Purification of the peptides was performed on an HPLC machine equipped with photodiode array detector that was coupled to an RP-18e reversed phase column (250 × 25 mm), using a similar gradient as for the analytical HPLC but with a flow rate of 20 mL min−1. The cleaved peptides were >90% pure (as inferred from HPLC), and were obtained as pure compounds after preparative HPLC. Analysis of peptide 1 (H–(Arg–Trp)3–Lys(C(O)CH2CH2CCH)–NH2): RP-HPLC (C18): tR = 17.50 min; ESI-MS: m/z 627.18 (calc. 627.26 for [M + H]2+). Peptide 2 (H–(Arg–Trp)3–N(H)CH2CCH): RP-HPLC (C18): tR = 17.6 min; ESI-MS: m/z 1082.20 (calc. 1082.59 for [M + H]+). Peptide 3 (FcC(O)–Trp–(Arg–Trp)2–N(H)CH2CCH): RP-HPLC (C18): tR = 20.3 min; ESI-MS: m/z 1138.08 (calc. 1138.48 for [M + H]+).
2.4. Redox-initiated graft polymerization
A commercial XLE RO membrane was cut into pieces (1 × 2 cm2), disinfected and washed with 50% (v/v) isopropyl alcohol (3 × 10 min) and with distilled water (3 × 10 min) in an ultra-sonication bath (Bandelin Sonorex, Allpax, Germany). Redox-initiated graft polymerization was performed by dissolving K2S2O5 (67 mg, 0.25 mmol) and K2S2O8 (55 mg, 0.25 mmol), each in 11.7 mL distilled water. Once dissolved, the solutions were added to an Erlenmeyer flask containing the membrane pieces and methacrylic acid (0.85 mL, final concentration 10 mM) and poly(ethylene glycol)methacrylate (average Mn 360) (0.81 mL, final concentration 2.5 mM) while stirring at 25 °C in a temperature-controlled water bath for three different time intervals (20 min, 30 min and 40 min). The membranes were then washed intensively with distilled water (3 × 15 min) in the ultra-sonication bath and then stored in distilled water at 5 °C.2.5. Attachment of azide linker
A solution of 1-amino-3-azidopropane (60 mM), EDC (20 mM) and NHS (20 mM) in PBS (5 mL, 0.1 M, pH 7.4) was added to six membrane samples of either the washed XLE commercial RO membranes or the grafted membranes in a reaction vial. The solution was agitated by a Unimax 1010 orbital platform shaker (Heidolph, Kelheim, Germany) overnight at room temperature (25 °C). The membranes were subsequently washed with PBS (3 × 10 min) and distilled water (3 × 10 min) with sonication and were stored in distilled water at 5 °C.2.6. Immobilization of peptides using click chemistry
In general, the copper catalyzed azide–alkyne cycloaddition was performed on an azide-functionalized membrane piece by adding to it solutions of peptides 1–3, CuSO4, and sodium ascorbate in distilled water for a total reaction volume of 0.5 mL. The reaction was performed with variable concentrations of each reagent, reaction temperatures, and reaction times. The membrane was subsequently washed two times with HCl (1.0 mM, 2 × 10 min) in order to remove absorbed Cu ions from the membrane (the membranes became colorless), and distilled water (2 × 10 min) in an ultra-sonication bath and stored in distilled water at 5 °C until further use.2.7. Membrane performance analysis
Flux, membrane permeability and salt rejection were determined using a custom made 200 mL cylindrical stirred (500 rpm) dead-end filtration cell with a total membrane area of 11.3 cm2. Flux measurements of three XLE RO membrane pieces were taken before and after treating the membranes at different temperatures (25 °C, 40 °C, 60 °C, 80 °C) for 20 min in an aqueous solution of cupric sulfate (1.0 mM) and sodium ascorbate (2.0 mM). A pressure of 10 bar was applied and the flux of pure water was determined by collecting and weighing the permeate every 5 min until the collected permeate reached a stable and constant flux value. Average flux was calculated before and after membrane treatment, and the percent flux reduction was calculated using (1 − (Fa/Fb)) × 100, where Fa is the permeate flux after treatment, and Fb is the permeate flux before treatment. For salt rejection measurements a NaCl-solution (2000 mg L−1, conductivity 3945 ± 20 μS cm−1) was used. After 10 min, the permeates of three test membranes were collected and the conductivity of the permeate was measured using an inoLab level 1 (WTW GmbH & Co., Weilheim, Germany). Since the salt concentration and solution conductivity are proportional, the salt rejection equation can also be expressed as a function of solution conductivity as ((Λf − Λp)/Λf) × 100, where Λf is the feed conductivity, and Λp is the permeate conductivity.2.8. Contact killing bioassay
A culture of E. coli was grown in LB (100 mL) at 37 °C until the turbidity of the mixture reached an optical density (OD) of 0.4 (600 nm). The bacteria were pelleted and a test inoculum containing 2 × 107 CFU mL−1 in sterile PBS was prepared. A membrane sample (2 cm2) was cut into pieces of approximately 2 mm2 and placed in an Eppendorf tube (2 mL). The membrane was sterilized with 70% ethanol (v/v) and then washed with PBS (3×), after which the E. coli test inoculum (0.5 mL) was added to the tube containing the test membrane. The tubes containing membrane and inoculum were centrifuged for 5 min at 14800 rpm and then vortexed for 10 s. This was repeated six times for a total bacterial incubation time of ca. 30 min. Finally the tube was ultra-sonicated35 in an ice-water bath for 30 s and viable bacteria, expressed in colony forming units (CFU mL−1), were counted on LB agar plates. The average of three experiments is reported including standard deviation.
3. Results and discussion
3.1. Preparation of peptides with azide functionality
Peptides with alkyne functionality (Fig. 1) were prepared. To that end, we used the recently developed SAM2-linker that, in contrast to the SAM1-linker,36 produces unprotected peptide side chains that have a free or a modified N-terminus and an acetylene-functionalized C-terminal carboxamide group.33 The peptides were synthesized employing standard Fmoc/tBu-based solid-phase peptide-synthesis procedures. They were then purified with preparative HPLC, and their identities were confirmed with mass spectrometry. Compared to other known AMPs, the peptides used are relatively short, namely five or six amino acids long. These peptides were selected in view of their potent broad spectrum antibacterial activity.34 They have been studied in great detail, and their mode of action has been established as delocalizing membrane-bound proteins.37 As such, they do not require to be internalized in bacteria before they can exert their antibacterial activity. Even more, due to their small size, variations can be introduced using synthetic methods in order to further enhance their antibacterial properties.38 Lastly, these short peptides can be prepared on a relatively large scale, allowing them to find widespread application as membrane coatings for anti-biofouling purposes.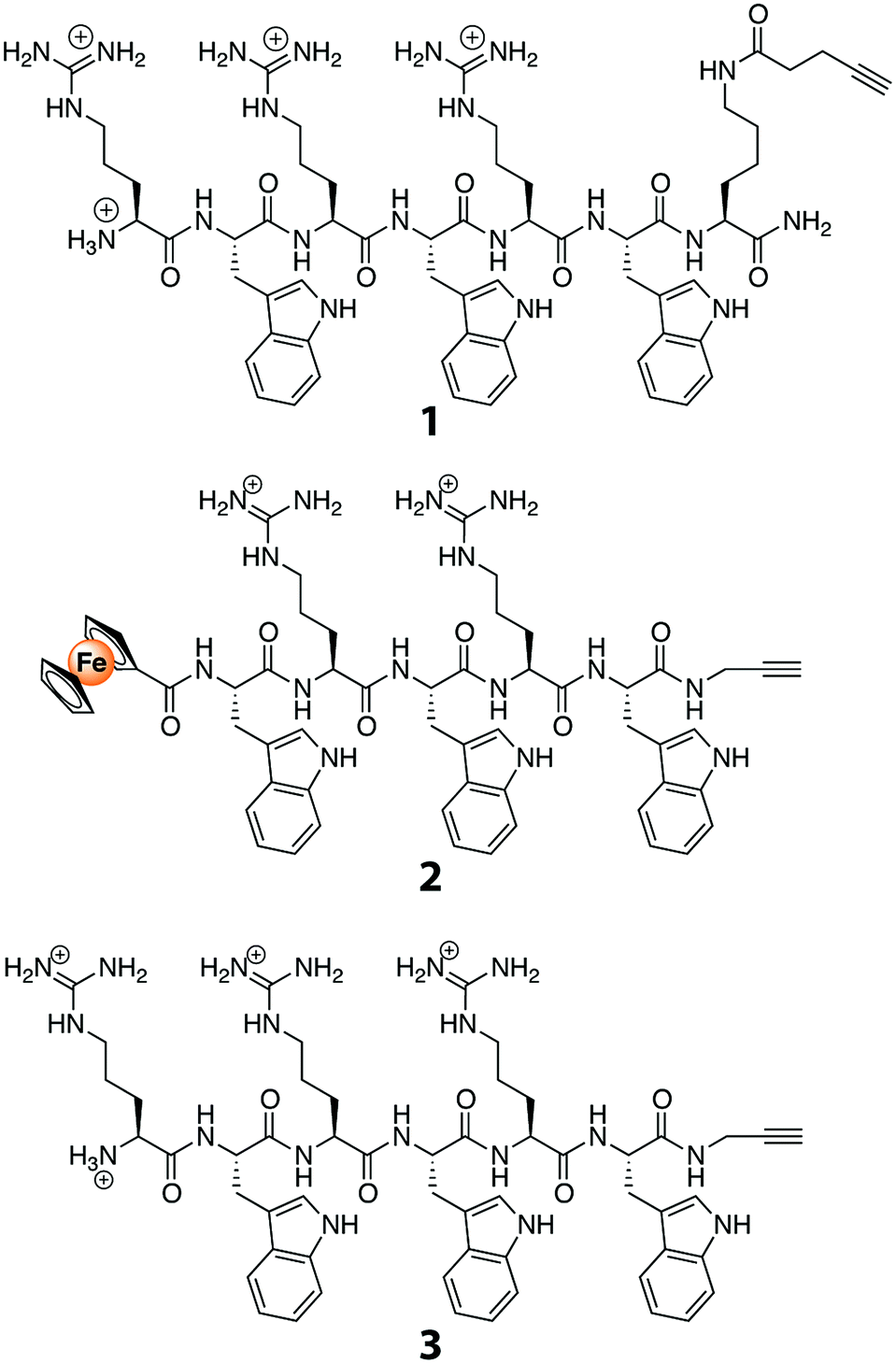 | ||
| Fig. 1 Structures of AMP alkynes used in the study. | ||
3.2. Grafting of TFC RO membranes
Azide functionalized membranes and alkyne functionalized peptides were prepared for this study. A commercial TFC RO membrane (Dow XLE) was used, in which the top layer was a thin film of aromatic polyamide polymer that includes surface exposed carboxylic acid moieties. To increase the amount of available carboxylic acid groups, a copolymer of methacrylic acid (MA) and poly(ethylene glycol)methacrylate (PEGMA) was grafted from the surface at a constant temperature of 25 °C using the redox radical initiators K2S2O5 and K2S2O8 (Fig. 2a).39–42 The reaction time was varied from 20 min to 40 min. The copolymer grafting reaction was monitored using FTIR spectroscopy by measuring the ratio of the signal measured at the spectral band corresponding to the carboxylic acid and ester carbonyl in the newly formed copolymer layer (1720 cm−1) to that measured at the band for the aromatic C–C signal of the polyamide surface (1488 cm−1). Signal ratios of 0.19, 0.78, and 0.97 were obtained for reaction times of 20 min, 30 min, and 40 min, respectively, and a reaction time of 30 min, which resulted in an intermediate amount of polymer on the membrane surface, was used to prepare the experimental membrane surfaces (Fig. 2b). Compared to the surface of the original membrane, this new membrane surface with increased carboxylic acid functionality was found to be more hydrophilic, with a water-drop contact angle of 37° compared to 57° on the original membrane. Both the grafted and non-grafted membranes were functionalized with an azide linker and used to test the optimization of peptide attachment.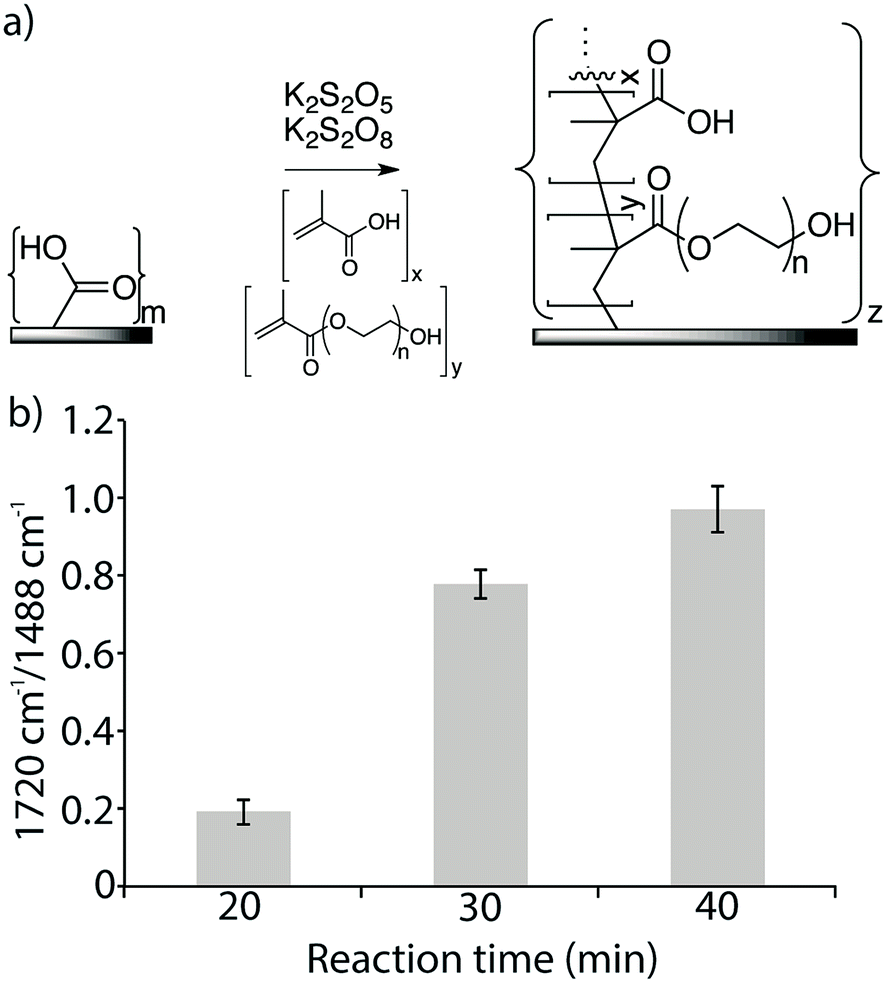 | ||
| Fig. 2 (a) LE 440 RO membrane surfaces were modified with a copolymer of methacrylic acid and poly(ethylene glycol)methacrylate. (b) FTIR absorbance ratio 1720 cm−1/1488 cm−1 of modified surfaces at different reaction times, at 25 °C. | ||
3.3. Coupling of antimicrobial peptides to the membrane
The azide linker 1-amino-3-azido-propane was immobilized on the membrane surface using EDC as the coupling reagent for a reaction time of 16 h to ensure efficient coupling (Fig. 3). Monitoring of the reaction using FTIR showed a new signal at 2100 cm−1, which indicated that the azide was covalently incorporated on the membrane surface. This signal was much stronger on the membrane-azide with the grafted copolymer layer compared to the azide on non-grafted membrane, suggesting that the grafted membrane had a higher amount of azide linker attached. In addition, azide immobilization led to further decreases of 16–19% in the contact angles of both the grafted and non-grafted membrane surfaces, indicating that their surfaces had greater hydrophilicity than an unaltered membrane.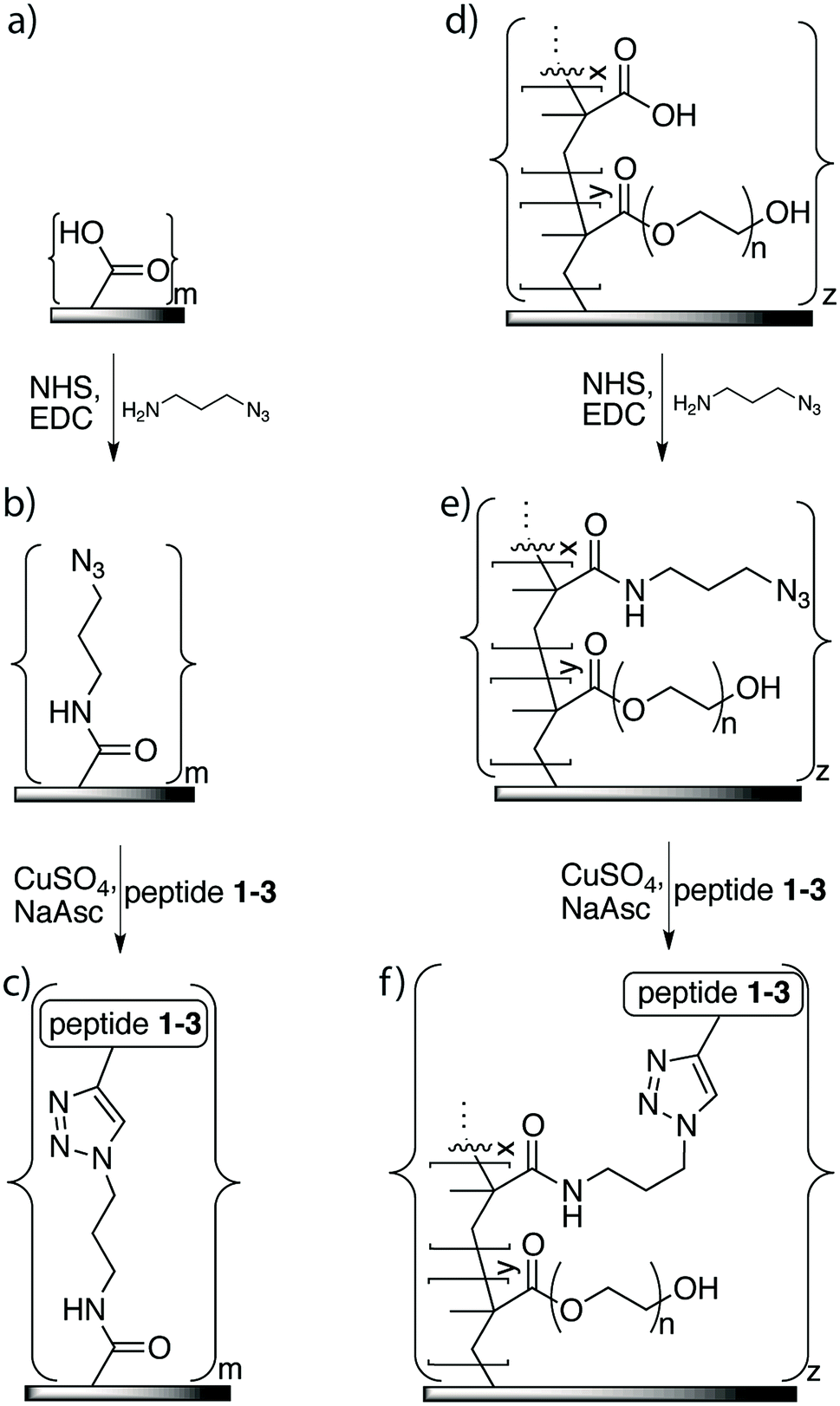 | ||
| Fig. 3 Attachment of antimicrobial peptides to LE 440 RO membrane surfaces: for non-grafted membrane surface (a): attachment of azide functionality (b); and immobilization of peptide using click chemistry (c). For membrane surface grafted with copolymer of methacrylic acid and poly(ethylene glycol)methacrylate (d): attachment of azide functionality to grafted surface (e); immobilization of peptide on grafted surface using click chemistry (f). | ||
The peptide immobilization reaction (Fig. 3) was optimized based on the following considerations: (i) the reaction time should be relatively short, (ii) it should consume minimum amounts of peptide and catalyst, and (iii) the reaction conditions should not adversely affect membrane performance. To this end, we systematically varied reaction time, temperature, amount of peptide, and amount of catalyst.
First, we varied the temperature from 25 °C to 80 °C and found, based on FTIR analysis of the azide-bands on the membranes, that the reaction at the highest temperature was the most efficient (Fig. 4a). The hydrophobicity also increased, which was indicated by an increase in contact angle, and it was found to correlate with increases in the amount of surface-immobilized peptide (Fig. 4b). The reaction at 80 °C for 20 min was deemed to have a higher efficiency compared to the same reaction time at lower temperatures and compared to the long reaction time of 16 h at a lower temperature (25 °C). We compared untreated membranes with a measured NaCl rejection of 92.7 ± 0.4% to the same membranes treated with the highest temperature reaction conditions (80 °C, 20 min) and observed no significant differences to the NaCl rejection (p > 0.29). At 25 °C and 40 °C no change in pure water flux was observed. Only minor changes in flux values resulted after treating the membrane under the highest temperature conditions (60 °C or 80 °C, 20 min): a slight decrease in flux was observed, 9% or 14% respectively, compared to the untreated membranes. Modification of membrane surfaces may lead to a reduction of permeability. This is especially observed in the case of graft polymerization on membrane surfaces where reported permeability reductions can be up to 40%.43 Thus the benefits gained in the surface property modification should be weighed against the decreased membrane permeability. On the other hand, membranes may be engineered to maintain a high flux and rejection character by choosing appropriate membrane substrates. For example, PA-TFC membranes with permeabilities in the nanofiltration range were nanostructured with a surface graft polymerization technique, to obtain membranes performance values comparable to commercially available RO membranes.44
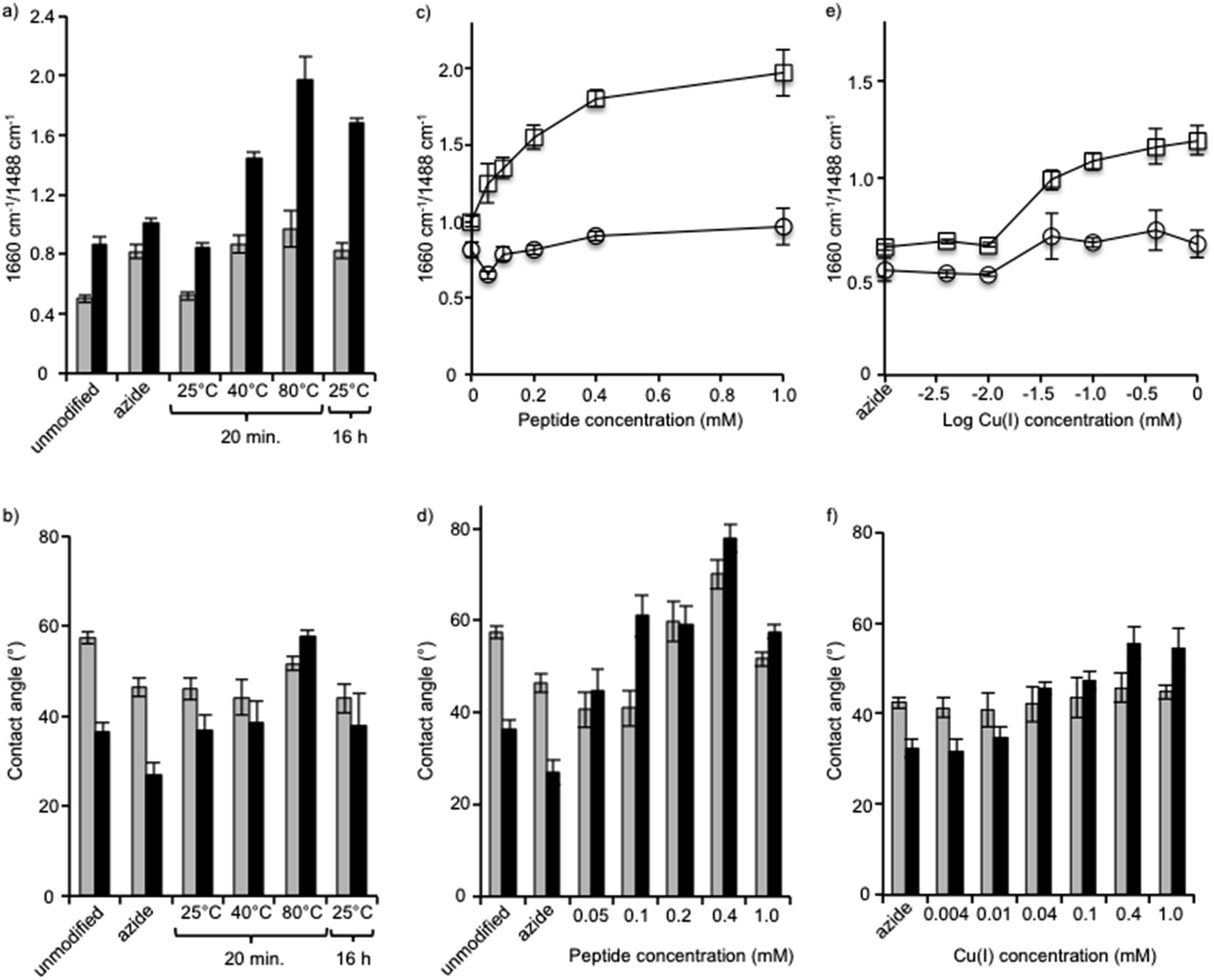 | ||
| Fig. 4 Effects of temperature (a and b), peptide concentration (c and d) and Cu(I) concentration (e and f) on peptide immobilization as shown by FTIR (a, c and e) and contact angle measurement (b, d and f). XLE RO membranes (gray, (○)), and grafted RO membranes (black, (□)). | ||
Next, using the short reaction time of 20 min at a temperature of 80 °C, the reaction was performed using a range of peptide concentrations from 0.05 mM to 1.0 mM (Fig. 4c and d). FTIR analysis of each membrane showed that the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/aromatic C–C intensity ratio steadily grew as the peptide concentration was increased from 0.05 mM to 0.4 mM, but from 0.4 mM to the highest concentration of 1.0 mM no significant change occurred. With the exception of the reaction with 1.0 mM peptide, in all reactions the membrane contact angle also increased proportionally to the AMP tethering efficiency, as seen by FTIR in which the contact angle was observed to be lower. Although a specific explanation for this phenomenon has not been offered, the fact that amphiphilic AMPs interact and aggregate when at high concentrations may influence the effect they have on the hydrophobicity of the membrane surface as measured by contact angle. Thus, FTIR may be a more reliable measure to determine the relative amounts of peptide on the membrane surface.
O/aromatic C–C intensity ratio steadily grew as the peptide concentration was increased from 0.05 mM to 0.4 mM, but from 0.4 mM to the highest concentration of 1.0 mM no significant change occurred. With the exception of the reaction with 1.0 mM peptide, in all reactions the membrane contact angle also increased proportionally to the AMP tethering efficiency, as seen by FTIR in which the contact angle was observed to be lower. Although a specific explanation for this phenomenon has not been offered, the fact that amphiphilic AMPs interact and aggregate when at high concentrations may influence the effect they have on the hydrophobicity of the membrane surface as measured by contact angle. Thus, FTIR may be a more reliable measure to determine the relative amounts of peptide on the membrane surface.
After identifying the optimal peptide concentration at 0.4 mM, the concentration of copper catalyst was varied from 0.004 mM to 1 mM using the short reaction time of 20 min at a temperature of 80 °C (Fig. 4e and f). In all cases, the copper(I) catalyst was generated in situ using a 1:2 ratio of CuSO4 and sodium ascorbate. The reaction, which proceeded only at copper concentrations of 0.04 mM and above, was seen clearly using both FTIR and contact angle measurements. The optimal reaction conditions, therefore, comprised the short reaction time of 20 min, a temperature of 80 °C, and concentrations of both peptide and copper of 0.4 mM.
Peptides 2 and 3 were applied to both the azide-XLE membranes and the azide-grafted copolymer-XLE membranes using the optimal coupling conditions identified above. After peptide application, the membrane surfaces were analyzed with X-ray photoelectron spectroscopy (XPS), a technique that gives quantitative information about the elemental composition of the surface to a depth of 5–10 nm. An anticipated indication of peptide attachment, especially for the polymer grafted membranes, can be observed in nitrogen content. We expected the grafted control membrane to lack nitrogen and the membranes with peptide coupling to show increased nitrogen content due to the presence of nitrogen in the peptide amides, the arginine residues, and the tryptophan residues. Indeed, we observed only 0.35% nitrogen in the control grafted membranes compared with 14–15% after peptide attachment.
Unique to peptide 2 was the presence of iron (Fe2+) in the form of a ferrocene derivative, which aided in membrane surface characterization and gave insight into the surface concentration of the covalently attached peptide. No detectable amounts of Fe2+ were observed in the control membranes or in membranes coupled with peptide 3 (Table 1). As expected, Fe2+ was only detected in the membranes coupled with peptide 2 at 0.35% and 0.45% for the un-grafted and the grafted membranes, respectively. This minor difference in Fe2+ revealed that the amount of peptide available on the surface was relatively similar for both membrane types. However, ATR-FTIR revealed larger differences in amide band absorption ratios (1660 cm−1/1488 cm−1) for the grafted compared to the un-grafted membranes (see Fig. 4), indicating that the peptide was incorporated throughout the grafted layer and not only on its surface. Since the AMPs are covalently bound to the polymer, a mechanism of leaching from within the polymer to contact bacteria is not possible. Thus an effective antimicrobial surface requires that the antimicrobial be present on the surface layer of the membrane to ensure physical contact with bacterial components. This would also ensure efficient use of the amount of AMP needed for modification. Development of effective antimicrobial surfaces via covalent attachment of AMPs will also depend on other factors such as peptide orientation,45 peptide structure and surrounding polymer chemistry,46 and other parameters such as tethering length.47
3.4. Antimicrobial surface activity
The antimicrobial effectiveness of the AMP-functionalized membranes was tested using a direct inoculum method that involved centrifugation of the bacteria on the membrane surfaces. The centrifugation step was added in order to enhance the probability of the bacteria to contact the membrane. To evaluate the bactericidal ability of the peptides upon contact with bacteria, the bacteria were incubated for a total time of approximately 30 min. For E. coli, a 32% and 36% decrease in CFU was observed for both peptides 2 and 3 compared to the unmodified, control membrane (Fig. 5). In comparison, the membranes with the peptide-copolymer coating showed less activity for either sequence. This finding indicates that tethered AMP activity is influenced by AMP surface immobilization parameters, i.e. surface concentration, spacer length, flexibility, and orientation of AMP, but also by membrane surface composition and properties to which the AMP is attached.48 Research to further elucidate these findings with respect to RO membrane surfaces is ongoing. The hexapeptide reported herein was ideal for method development, and facilitates future surface antimicrobial activity studies with a series of AMPs that will be reported in due course.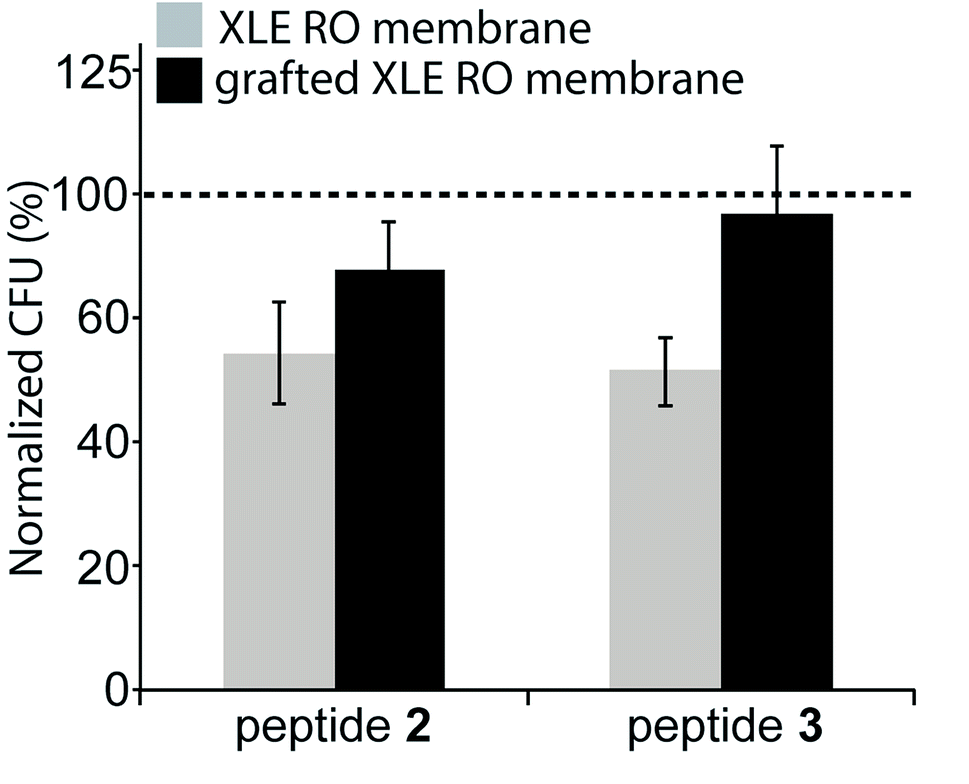 | ||
| Fig. 5 Antimicrobial effect of peptides bonded to the membranes: E. coli colony forming units (CFU) after 30 min exposure, as percentage of CFU on unmodified membranes controls. Two types of control membranes are used and indicated with the dashed line: commercial XLE membrane (control for gray bars), or the copolymer coated XLE membrane surface (control for the black bars). Average of 3 independent experiments including the standard deviation is shown. | ||
4. Conclusions
The aim of this study was to optimize the reaction conditions for antimicrobial peptide immobilization on RO membrane surfaces while maintaining membrane performance. Peptides that were directly coupled to the membrane surface showed increased antimicrobial activity compared to peptides on membrane surfaces that were modified with a copolymer. This study provides the necessary tools for the efficient attachment of a wide variety of antimicrobial and other compounds to RO membrane surfaces. In addition, it may contribute to an overall better understanding of the antibacterial and potential anti-biofilm effects of tethered antimicrobial agents on RO membranes and on other surfaces.Acknowledgements
This research was supported by THE ISRAEL SCIENCE FOUNDATION (grant No. 1474-13) to CJA. EB thanks the German Ministry for Education and Research (BMBF) for funding his stay in Israel in the framework of the Young Scientists Exchange Program (YSEP). NK is grateful to the Pratt Foundation for a PhD Scholarship (Ben-Gurion University of the Negev).References
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- A. D. Khawaji, I. K. Kutubkhanah and J.-M. Wie, Desalination, 2008, 221, 47–69 CrossRef CAS.
- R. J. Petersen, J. Membr. Sci., 1993, 83, 81–150 CrossRef CAS.
- H.-C. Flemming, Exp. Therm. Fluid Sci., 1997, 14, 382–391 CrossRef CAS.
- N. Misdan, W. J. Lau and A. F. Ismail, Desalination, 2012, 287, 228–237 CrossRef CAS.
- Q. Li and M. Elimelech, Environ. Sci. Technol., 2004, 38, 4683–4693 CrossRef CAS PubMed.
- H.-C. Flemming, T. Griebe, G. Schaule, J. Schmitt and A. Tamachkiarowa, Desalination, 1997, 113, 215–225 CrossRef CAS.
- P. Gilbert and M. R. W. Brown, in Microbial Biofilms, ed. H. E. Lappin-Scott and J. W. Costerton, Cambridge University Press, Cambridge, 1995, pp. 118–132 Search PubMed.
- Y. H. An, B. K. Blair, K. L. Martin and R. J. Friedman, in Handbook of Bacterial Adhesion, ed. Y. H. An and R. J. Friedman, 2000, pp. 609–625 Search PubMed.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- I. J. Roh, A. R. Greenberg and V. P. Khare, Desalination, 2006, 191, 279–290 CrossRef CAS.
- M. S. Rahaman, H. Thérien-Aubin, M. Ben-Sasson, C. K. Ober, M. Nielsen and M. Elimelech, J. Mater. Chem. B, 2014, 2, 1724–1732 RSC.
- J. Nikkola, X. Liu, Y. Li, M. Raulio, H.-L. Alakomi, J. Wei and C. Y. Tang, J. Membr. Sci., 2013, 444, 192–200 CrossRef CAS.
- K. R. Zodrow, M. E. Tousley and M. Elimelech, J. Membr. Sci., 2014, 453, 84–91 CrossRef CAS.
- L. Segev-Zarko, R. Saar-Dover, V. Brumfeld, M. L. Mangoni and Y. Shai, Biochem. J., 2015, 468, 259–270 CrossRef CAS PubMed.
- F. Costa, I. F. Carvalho, R. C. Montelaro, P. Gomes and M. C. L. Martins, Acta Biomater., 2011, 7, 1431–1440 CrossRef CAS PubMed.
- I. E. Ivanov, A. E. Morrison, J. E. Cobb, C. A. Fahey and T. A. Camesano, ACS Appl. Mater. Interfaces, 2012, 4, 5891–5897 CAS.
- K. Berliner, E. Hershkovitz, Z. Ronen and R. Kasher, in Peptides for Youth, ed. S. DelValle, E. Escher and W. D. Lubell, Springer Science+Business Media, 2009, pp. 241–242 Search PubMed.
- C. J. Arnusch, A. M. J. J. Bonvin, A. M. Verel, W. T. M. Jansen, R. M. J. Liskamp, B. de Kruijff, R. J. Pieters and E. Breukink, Biochemistry, 2008, 47, 12661–12663 CrossRef CAS PubMed.
- C. J. Arnusch, H. Branderhorst, B. de Kruijff, R. M. J. Liskamp, E. Breukink and R. J. Pieters, Biochemistry, 2007, 46, 13437–13442 CrossRef CAS PubMed.
- C. J. Arnusch, R. J. Pieters and E. Breukink, PLoS One, 2012, 7, e39768 CAS.
- X.-M. Wu, L.-L. Wang, Y. Wang, J.-S. Gu and H.-Y. Yu, J. Membr. Sci., 2012, 421, 60–68 CrossRef.
- R. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217–6223 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. van Scherpenzeel, M. van der Pot, C. J. Arnusch, R. M. J. Liskamp and R. J. Pieters, Bioorg. Med. Chem. Lett., 2007, 17, 376–378 CrossRef CAS PubMed.
- I. E. Valverde, A. Bauman, C. A. Kluba, S. Vomstein, M. A. Walter and T. L. Mindt, Angew. Chem., Int. Ed., 2013, 52, 8957–8960 CrossRef CAS PubMed.
- H.-Y. Yu, Y. Kang, Y. Liu and B. Mi, J. Membr. Sci., 2014, 449, 50–57 CrossRef CAS.
- H. B. Albada, P. Prochnow, S. Bobersky, S. Langklotz, P. Schriek, J. E. Bandow and N. Metzler-Nolte, ACS Med. Chem. Lett., 2012, 3, 980–984 CrossRef CAS PubMed.
- M. Strack, N. Metzler-Nolte and H. B. Albada, Org. Lett., 2013, 15, 3126–3129 CrossRef CAS PubMed.
- H. B. Albada, A.-I. Chiriac, M. Wenzel, M. Penkova, J. E. Bandow, H.-G. Sahl and N. Metzler-Nolte, Beilstein J. Org. Chem., 2012, 8, 1753–1764 CrossRef CAS PubMed.
- M.-Y. Lutskiy, S. Avneri-Katz, N. Zhu, M. Itsko, Z. Ronen, C. J. Arnusch and R. Kasher, Sep. Purif. Technol., 2015, 141, 214–220 CrossRef CAS.
- M. Strack, S. Langklotz, J. E. Bandow, N. Metzler-Nolte and H. B. Albada, J. Org. Chem., 2012, 77, 9954–9958 CrossRef CAS PubMed.
- M. Wenzel, A. I. Chiriac, A. Otto, D. Zweytick, C. May, C. Schumacher, R. Gust, H. B. Albada, M. Penkova, U. Krämer, R. Erdmann, N. Metzler-Nolte, S. Straus, D. Becher, H. Brötz-Oesterhelt, H.-G. Sahl and J. E. Bandow, Proc. Natl. Acad. Sci., 2014, 111, E1409–E1418 CrossRef CAS PubMed.
- H. B. Albada, P. Prochnow, S. Bobersky, S. Langklotz, J. E. Bandow and N. Metzler-Nolte, Chem. Sci., 2014, 5, 4453–4459 RSC.
- M. Herzberg, A. Sweity, M. Brami, Y. Kaufman, V. Freger, G. Oron, S. Belfer and R. Kasher, Biomacromolecules, 2011, 12, 1169–1177 CrossRef CAS PubMed.
- I. Eshet, V. Freger, R. Kasher, M. Herzberg, J. Lei and M. Ulbricht, Biomacromolecules, 2011, 12, 2681–2685 CrossRef CAS PubMed.
- S. Belfer, Y. Purinson and O. Kedem, Acta Polym., 1998, 49, 574–582 CrossRef CAS.
- S. Belfer, Y. Purinson, R. Fainshtein, Y. Radchenko and O. Kedem, J. Membr. Sci., 1998, 139, 175–181 CrossRef CAS.
- R. Bernstein, S. Belfer and V. Freger, Environ. Sci. Technol., 2011, 45, 5973–5980 CrossRef CAS PubMed.
- N. H. Lin, M. Kim, G. T. Lewis and Y. Cohen, J. Mater. Chem., 2010, 20, 4642–4652 RSC.
- Y. Li, S. Wei, J. Wu, J. Jasensky, C. Xi, H. Li, Y. Xu, Q. Wang, E. N. G. Marsh, C. L. Brooks III and Z. Chen, J. Phys. Chem. C, 2015, 119, 7146–7155 CAS.
- K. Yu, J. C. Lo, Y. Mei, E. F. Haney, E. Siren, M. T. Kalathottukaren, R. E. Hancock, D. Lange and J. N. Kizhakkedathu, ACS Appl. Mater. Interfaces, 2015, 7, 28591–28605 Search PubMed.
- L. D. Lozeau, T. E. Alexander and T. A. Camesano, J. Phys. Chem. B, 2015, 119, 13142–13151 CrossRef CAS PubMed.
- S. A. Onaizi and S. S. J. Leong, Biotechnol. Adv., 2011, 29, 67–74 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |