DOI:
10.1039/C6RA21900D
(Paper)
RSC Adv., 2016,
6, 99729-99734
Nanosupramolecular assembly of amphiphilic guest mediated by cucurbituril for doxorubicin delivery†
Received
1st September 2016
, Accepted 10th October 2016
First published on 12th October 2016
Abstract
Nanosupramolecular amphiphilic assemblies have aroused significant attention in the biomaterials field and find potential applications in biomimetic synthesis, controlled/sustained drug release, and targeted imaging and diagnosis. In this work, a drug delivery system has been constructed by the host–guest complexation of cucurbit[6]uril (CB[6]) with a bifunctional guest compound (DTA) possessing one alkyl head and two ammonium tails. The assistance of CB[6] can dramatically decrease the critical aggregation concentration of this amphiphilic guest, thus leading to the formation of uniform and stable supramolecular nanoparticles (DTA·CB[6] NPs) at relatively lower concentration in water. Spectroscopic and microscopic experiments jointly demonstrate that the obtained NPs can not only encapsulate hydrophobic dye as molecular probe but also load hydrophilic drug (doxorubicin, DOX) in its internal microenvironment. In addition, when the hydrophobic region of DTA is included by α-cyclodextrin, the DTA·CB[6] NPs are completely dissipated and the encapsulated substrates can be released in a controlled manner. More interestingly, the DOX-loaded NPs exhibited better anticancer activity toward tumor cells, and therefore, the present study may provide a biocompatible platform in the construction of smart nanocarriers for on-demand drug delivery and release.
Introduction
In the field of supramolecular chemistry, the concept of “supra-amphiphiles”, multicomponent amphiphiles constructed by amphiphilic guests and macrocyclic hosts through synergistic noncovalent interactions, has provided an alternative and powerful strategy in the fabrication of smart biofunctional nanocarriers, mainly due to their immense advantages of easier maneuverability, better bioavailability, and more accessible stimuli-responsive capability.1 Among various macrocyclic receptors, cucurbit[n]urils (CB[n], n = 5–10), a family of cyclic compounds consisting of one hydrophobic cavity and two carbonyl-laced portals to complex organic cations in water, have been explored as a promising candidate in the construction of amphiphilic supramolecular nanosystems, because CBs gather the following structural and molecular binding features for controlled delivery and release: (1) directly encapsulating positively charged guests originating from their cation-receptor properties, which has complementary superiorities to other classic macrocycles (e.g. cyclodextrin);2 (2) enhancing the bioavailability of protonated guests by CB-induced pKa shift, which can further regulate the charge density of host–guest complexes;3 and (3) increasing the included guest's chemical stability and reactivity via macrocyclic protection.4 Thus, it is believed that the rational design of CB-based supra-amphiphiles will be developed into a new approach to efficiently transport administrated drug molecules.
In previous studies, it has been demonstrated that many ionized macrocycles, such as sulfonated calixarenes and cyclodextrins, can induce molecular aggregation behaviours and lower critical aggregation concentration (CAC) mainly through electrostatic interactions.5 Researchers have exploited macrocycle-induced aggregation to construct supramolecular binary micelles and vesicles, which can be utilized as advanced drug delivery vehicles with responsive capability to various internal and external stimuli. However, in contrast to these known supra-amphiphilic systems, the research on the molecular aggregation behaviours using neutral CBs still deserve our careful attention, and therefore, it seems imperative to explore the intermolecular binding features and potential biomedical applications of CB-involved amphiphilic nanoassemblies.6 Inspired by the fascinating reported results, in this work, we synthesized an amphiphilic molecule bearing two ammonium sidechains as the hydrophilic head and one dodecyl group as the hydrophobic tail, which could form nanoparticles (NPs) at a relatively higher concentration. Remarkably, it is found that the CAC value could dramatically decrease upon addition of CB[6], which were comprehensively verified by spectroscopic and microscopic experiments. Meanwhile, the assembling and disassembling of supramolecular NPs could be conveniently regulated by CB[6] and α-cyclodextrin (α-CD). Moreover, the obtained supramolecular NPs formed by the binary inclusion complex could entrap both hydrophobic dye and hydrophilic drug (Scheme 1). Consequently, the drug-loaded NPs gave improved anticancer activity toward tumor cells. The CB-involved superamphiphile in the present study may find practical possibilities for the development of novel drug delivery systems.
 |
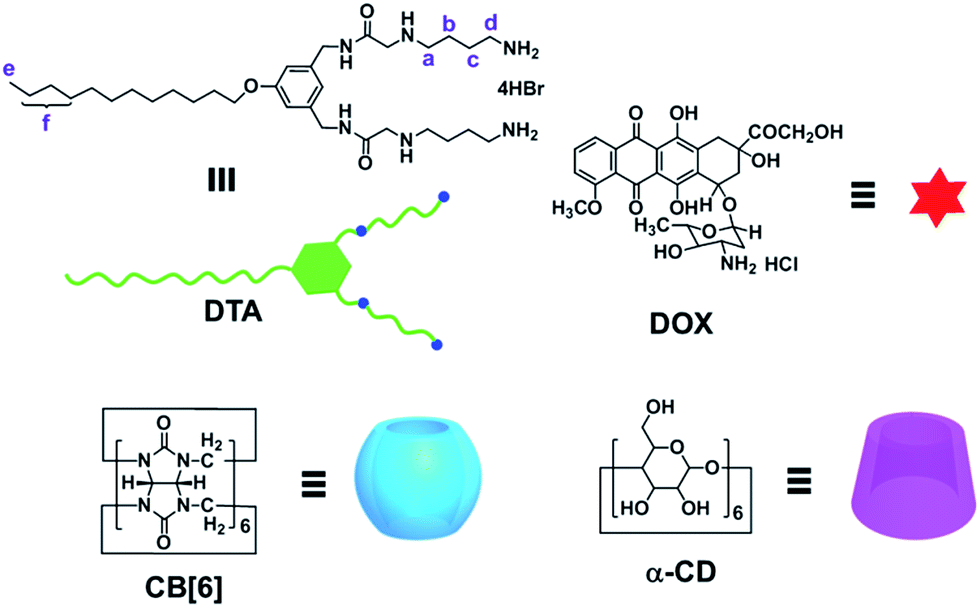
| | Scheme 1 Chemical structures of DTA, DOX, CB[6] and α-CD. | |
Experimental
Synthesis of amphiphilic guest DTA
Compound 5 (200 mg) was dissolved in the solution of HBr–CH3COOH (10 mL), and the mixture was stirred at room temperature for 20 h. Then diethyl ether (50 mL) was added into the mixture, and white solid was produced at the same time. After centrifugation, white solid was collected to give guest molecule DTA (153 mg, 95%). 1H NMR (400 MHz, D2O) δ 6.84 (m, 3H), 4.37 (m, 4H), 4.05 (s, 2H), 3.92 (s, 4H), 3.09 (t, J = 8.0 Hz, 4H), 3.00 (t, J = 8.0 Hz, 4H), 1.74 (m, 11H), 1.23 (m, 20H), 0.81 (t, J = 4.0 Hz, 3H). 13C NMR (100 MHz, D2O) δ 165.6, 158.9, 139.4, 117.9, 112.7, 68.2, 48.4, 47.2, 43.1, 39.0, 31.9, 29.8, 29.6, 29.4, 29.3, 26.1, 24.1, 22.9, 22.7, 14.0. HRMS (MALDI): m/z: 577.4805 [M − 3HBr − Br]+; 599.4623 [M − 4HBr + Na]+.
Preparation the DTA@CB[6] NPs
DTA (1.89 mg, 0.7 mM) and CB[6] (4.18 mg, 1.4 mM) were dissolved at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molecular ratio in 3 mL Milli-Q water. The solution was sonicated for 30 min and then filtered through a membrane filter (pore size: 450 nm, Millipore) to obtain the binary nanoparticles.
:2 molecular ratio in 3 mL Milli-Q water. The solution was sonicated for 30 min and then filtered through a membrane filter (pore size: 450 nm, Millipore) to obtain the binary nanoparticles.
Preparation DOX-loaded DTA@CB[6] NPs
DOX-loaded nanoparticles were prepared by dissolving DTA (6.3 mg), CB[6] (14 mg) and DOX (2 mg) in 10 mL of Milli-Q water. Then the mixed solution was stirred for 24 h at room temperature in darkness. The resulting solution was dialyzed (MW cutoff 3500) for 24 h to remove the free DOX. The DOX concentration was determined by its absorption at 490 nm by UV/vis spectroscopy. Moreover, the drug encapsulation and loading efficiencies can be calculated using the following equations:
| Encapsulation efficiency = 100% × mdrug in nanoparticles/mtotal drug = 100% × 1.10 mg/2.0 mg = 55.0% |
| Drug loading content = 100% × mdrug in nanoparticles/mnanoparticles = 100% × 1.10 mg/20.3 mg = 5.4% |
In vitro release of DOX from DTA@CB[6] NPs with α-CD
To investigate if DOX could be slowly released from DTA@CB[6] NPs, free DOX solution and the DOX-loaded DTA@CB[6] NPs solution with and without α-CD were prepared. Then each sample solution (3 mL, [DOX] = 0.33 mg mL−1) dissolved in PBS (pH = 7.2, I = 0.01 M) was placed in a dialysis membrane (MW cutoff 3500) and tightly sealed, which was immersed into 200 mL PBS in a beaker in the dark at 37 °C. The release percentage data were collected after the DOX-loaded NPs were immersed in the dialysis membrane for 30 min. At selected time intervals, 3 mL sample was taken out from buffer solution outside the dialysis bag and supplemented with 3 mL fresh buffer solution. The amount of released DOX was monitored by measuring its UV absorbance at 490 nm.
In vitro cytotoxicity assay
Human breast cancer cells (MCF-7) and mouse embryonic fibroblasts (NIH3T3) were purchased from the Peking Union Medical College Hospital (PUMCH, Beijing, China). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from the Dingguo Biotechnology Company (Tianjin, China). MCF-7 and NIH3T3 cells were cultured in DMEM, which was supplemented with 10% FBS, in 96-well plates (4 × 104 cells per mL, 0.1 mL per well) at 37 °C under a humidified atmosphere with CO2 (5%) for 24 h. Next, the cells were incubated with culture medium containing DOX, DTA@CB[6] NPs, and DOX-loaded NPs ([DOX] = 1.8 μM, [DTA] = 7 μM, and CB[6] = 14 μM). After incubation for 24 h, 50 μL culture medium was removed and 50 μL of 5 mg mL−1 MTT solution in PBS were added into each well. After incubation for 4 h, all medium were removed carefully. Then the blue formazan crystals were dissolved by 100 μL dimethyl sulfoxide, and the absorbance at a wavelength of 490 nm of each well was collected. Differences among the treatment groups were statistically analysed using the paired Student's t-test. A statistically significant difference was reported if p < 0.05.
Results and discussion
Construction of binary DTA·CB[6] nanoparticles
The molecular binding behaviours of DTA with CB[6] and α-CD were investigated by 1H NMR spectroscopy in D2O. As shown in Fig. 1, the characteristic peaks of the DTA's diamine chains (Ha–d) significantly shifted to the upper field in the presence of CB[6], corroborating the formation of highly stable host–guest complex between butanediamine and CB[6].7 Conversely, the dodecyl protons (He–f) displayed pronounced complexation-induced down field shifts, mainly due to the hydrophobic interaction between aliphatic chain and α-CD's cavity. Moreover, when DTA was mixed with α-CD and CB[6] together, the overall NMR spectral characteristics resembled the sum of the chemical shift changes in the case of DTA·CB[6] and DTA·α-CD complexes (Fig. 1). These contrasting NMR chemical shifts verified that the pendant diammonium and dodecyl chains in DTA could be specifically bound to CB[6] and α-CD, which provided us with non-interfering binding sites to regulate the NP's morphology, as described below.
 |
| | Fig. 1 1H NMR spectra (400 MHz, D2O, 25 °C) of (a) DTA + CB[6] + α-CD, (b) DTA + α-CD, (c) DTA, (d) DTA + CB[6] ([DTA] = [α-CD] = 0.5 mM and [CB[6]] = 1.0 mM). | |
Possessing four positive charges as hydrophilic head and a long dodecyl chain as hydrophobic tail, the amphiphilic DTA could self-aggregate into spherical nanoparticles above its CAC value.8 First, DTA's CAC was measured by monitoring the dependence of electrical conductivity by varying the DTA concentration. As can be seen from Fig. 2a, the electrical conductivity was sharply increasing with the increase of DTA concentration, and there was a point of inflection at 13 mM, indicative of the formation of large-sized aggregates in solution. Comparatively, when the hydrophilic head of DTA was occupied by 2 equiv. of CB[6], it is found that the optical transmittance at 400 nm gradually decreased as the concentration of DTA·CB[6] assembly increased, accompanied by the an inflection point was observed at 0.7 mM assigned to the CAC value of DTA·CB[6] system, which was 19 times lower than the one of free DTA (Fig. 2b). The CB-enhanced molecular aggregation was also confirmed by the electrical conductivity experiment, from which a similar CAC value was obtained (see Fig. S13 in ESI†). In our case, the macrocycle-assisted binding behaviours in DTA·CB[6] assembly were definitely attributed to the strong supramolecular complexation of CB[6] with cationic diammonium sites in DTA. That is, the unfavourable electrostatic repulsion among the intensive positive charges of DTA can be greatly eliminated through the N+⋯Oδ− ion-dipole interconnection between DTA's ammonium sites and CB[6]'s carbonyl groups and meanwhile, the encapsulation with bulky CB[6] can increase the hydrophilic performance of DTA·CB[6] assembly to a large extent, thus leading to the formation of supra-amphiphiles at a relatively lower concentration.
 |
| | Fig. 2 (a) Dependence of electrical conductivity on the DTA concentration and (b) dependence of the optical transmittance at 400 nm on the concentration of DTA·CB[6] assembly in water ([CB[6]] = 2[DTA], 25 °C, pH 7.0). In transmittance measurements, the optical signals were monitored at long-wavelength region (400 nm) to completely avoid any absorption of free DTA or CB[6]. | |
Next, the aggregation morphology was explored in both solution and the solid state. It was found that the DTA·CB[6] spherical NPs possessed an average diameter of 70 nm in the scanning electron microscopic (SEM) and transmission electron microscopic (TEM) images. Along with these microscopic investigations, dynamic light scattering (DLS) results showed that the average diameter of DTA·CB[6] assembly was 63 nm, which was basically consistent with TEM and SEM results. Meanwhile, a clear Tyndall effect was observed for a solution of binary DTA·CB[6] complex, but no such phenomenon was found for free DTA under the same experimental condition, here again demonstrating the complexation-induced aggregation in solution (Fig. 3). Moreover, the zeta potential of the DTA·CB[6] NPs was obtained as +32.88 mV, which was mainly attributed to the peripheral ammonium units of butanediamine tails in DTA (see Fig. S14 in ESI†).
 |
| | Fig. 3 (a) DLS results of DTA·CB[6] NPs; (b) Tyndall effect of (i) DTA·CB[6] NPs and (ii) free DTA ([DTA] = 0.7 mM and [CB[6]] = 1.4 mM); (c) TEM and (d) SEM images of the obtained DTA·CB[6] NPs. | |
Dye encapsulation by DTA·CB[6] NPs
In order to verify the encapsulation capability of DTA·CB[6] NPs, a hydrophobic fluorescence dye, nile red (NR), was used as molecular probe. As shown in Fig. 4, the fluorescence of NR at 630 nm was much weaker when dissolved in 5% methanol–water mixed solvent. However, the emission intensity was enhanced when NR was introduced into the aqueous solution of DTA·CB[6] NPs. These results indicated that NR could be readily incorporated into the hydrophobic domain of DTA·CB[6] nanoaggregates. In addition, control experiments also showed that DTA alone could not induce an obvious fluorescent enhancement of NR without assistance of CB[6], further suggesting the power of supramolecular complexation in the formation of amphiphilic nanostructures.
 |
| | Fig. 4 Fluorescence intensity of NR and NR-loaded NPs in the absence and presence of α-CD in water at 25 °C (λex = 530 nm, [NR] = 1.0 μM, [DTA] = 0.7 mM, [CB[6]] = 1.4 mM, and [α-CD] = 0.7 mM). | |
Considering that the alkyl chain and α-CD could from 1:1 stable host–guest complex with moderate binding constant of 103 M−1 order of magnitude, α-CD was used to modulate the disaggregation of the binary DTA·CB[6] NPs.9 As expected, when alkyl chain was included by α-CD, the hydrophile–lipophile balance would be broken, and thus the NPs would be dissipated. Accordingly, the fluorescent intensity of NR was instantly reduced upon addition of excess amount of α-CD, which indicated that NR was exposed into aqueous phase after the inclusion complexation of the DTA's dodecyl moiety with α-CD's cavity.
Drug loading and stimuli-responsive release behaviours of DTA·CB[6] NPs
After validating that the obtained NPs could be disaggregated with biocompatible α-CD, it might be suitable to construct smart drug delivery system using ternary DTA·CB[6]·α-CD complex. Subsequently, doxorubicin (DOX) was chosen as a fluorescent model compound to examine the drug loading and stimuli-responsive release behaviours. As compared to the original DTA·CB[6] NPs, the DOX-loaded ones gave an absorbance peak at 490 nm, which was the characteristic absorption of DOX in water. More intuitively, the colour of DOX-loaded NPs turned from colourless to light pink after dialysis, further indicating the successful entrapment of DOX into the DTA·CB[6] cores (Fig. 5a). Moreover, as derived from the UV/vis spectroscopic titrations, the drug encapsulation and loading efficiencies were measured to be 55.0% and 5.4%, respectively. Meanwhile, the morphology of DOX-loaded NPs was studied by TEM and DLS experiments (Fig. 5b and c). The results indicated that the diameter of DOX-loaded NPs was around 100 nm, which was larger than the unloaded NPs. When α-CD was added to the solution of DOX-loaded NPs, the NPs were disaggregated and amorphous structure was found in TEM images (Fig. 5d). Moreover, as shown in the drug release profiles, the addition of α-CD could dramatically accelerate the drug release rate, whereas the NPs-protected DOX gave much slower release behaviour in buffer solution. Accordingly, the release rate could be obtained as 32 μg h−1 for free DOX, 38 μg h−1 for DOX-load NPs with α-CD, and 26 μg h−1 for DOX-load NPs without α-CD, respectively. Meanwhile, the drug release percentage was 80% for free drug and 65% for NPs, respectively, after 4 h (Fig. 6). The sustained release of drug molecules may be beneficial for prolonging the retaining time in cellular environment and then reducing the adverse effect of original DOX.10
 |
| | Fig. 5 (a) UV-vis absorption spectrum of NPs and DOX-loaded NPs. Inset: colour changes of DOX-loaded NPs (right) and NPs (left); (b) DLS results of DOX-loaded NPs; TEM images of DOX-loaded NPs in the (c) absence and (d) presence of α-CD. (e) The assembling process of DOX-loaded NPs. | |
 |
| | Fig. 6 (a) In vitro release profiles of DOX from the DOX-loaded NPs in absence and presence α-CD in phosphate buffer solution (pH = 7.2, I = 0.01 M) at 37 °C. (b) The disassembling process of DOX-loaded NPs with α-CD. | |
In vitro cytotoxicity experiments
Finally, cytotoxicity experiments were carried out to evaluate the antitumor activity of the DOX-loaded NPs in vitro. Human breast cancer cells (MCF-7) and mouse embryonic fibroblasts (NIH3T3) were selected as the model cell lines. The DTA·CB[6] NPs showed no obvious cytotoxicity towards normal cells, indicating our obtained NPs were basically biocompatible. As shown in Fig. 7, DOX-loaded NPs displayed a better anticancer activity than free DOX toward MCF-7 cancer cells after 24 h incubation. The relative cellular viability of MCF-7 for DOX-loaded NPs was 35.7%, which was relatively lower than the one for free drug (60.9%). Meanwhile, it was found that the cell morphological characteristics were also consistent with results obtained from the aforementioned cytotoxicity experiments. These phenomena may be contributed to the self-assembled DTA·CB[6] NPs with suitable size and multivalent loading capacity for sustained drug release.11 In addition, the complexation of butanediamine with CB[6] could make positive pKa shift of around 1.0 unit and thus enhance the cation density of the whole NPs, which may also facilitate the internalization process through cell membrane.12
 |
| | Fig. 7 Relative cellular viability of (a) MCF-7 and (f) NIH3T3 cell lines after the treatment with DOX-loaded NPs, DOX, and NPs, respectively, after 24 h incubation. And cell photos of (b–e) MCF-7 and (g–j) NIH3T3 cell lines after the treatment with blank (b and g), DOX-loaded NPs (e and h), DOX (d and i), and NPs (e and j), respectively ([DOX] = 1.8 μM). The statistically significant differences were indicated with asterisks (*p < 0.05). | |
Conclusions
In conclusion, by utilizing the complexation-assisted aggregation strategy, we have constructed a drug delivery system by the noncovalent association of alkyl chain-modified polyamine with biocompatible CB[6]. Remarkably, the critical aggregation concentration of synthetic amphiphilic ammonium salt pronouncedly decreased by about 19 times owing to the complexation of CB[6]. Moreover, by benefiting from the high molecular binding affinity between dodecyl group and α-CD, the disassembling of NPs can be triggered by the addition of α-CD and the encapsulated drug molecules can be readily released in a controlled manner. More gratifyingly, the in vitro experiments further indicated that the DOX-loaded nanoassembly exhibited better anticancer activity toward MCF-7 cancer cells, but was safe to normal cells. Thus, we can envision that the present study may promote the CB-based supra-amphiphiles as intelligent biomaterials to enhance the drug loading efficiency and minimize the side effects in oncological therapy.
Acknowledgements
We thank the National Natural Science Foundation of China (No. 91227107, 21432004, and 21472100) for financial support.
Notes and references
-
(a) C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed;
(b) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
-
(a) S. J. Barrow, S. Kasera, M. J. Rowland, J. Del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed;
(b) N. Basilio, L. García-Río, J. A. Moreira and M. Pessêgo, J. Org. Chem., 2010, 75, 848–855 CrossRef CAS PubMed;
(c) E. Masson, X. Ling, R. Joseph and X. Lua, RSC Adv., 2012, 2, 1213–1247 RSC;
(d) B. C. Pemberton, R. K. Singh, A. C. Johnson, S. Jockusch, J. P. Da Silva, A. Ugrinov, N. J. Turro, D. K. Srivastava and J. Sivaguru, Chem. Commun., 2011, 47, 6323–6325 RSC;
(e) B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem.–Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed;
(f) B. C. Pemberton, N. Barooah, D. K. Srivatsava and J. Sivaguru, Chem. Commun., 2010, 46, 225–227 RSC.
-
(a) N. Barooah, J. Mohanty, H. Pal and A. C. Bhasikuttan, Proc. Natl. Acad. Sci., India, Sect. A, 2014, 84, 1–17 CrossRef CAS;
(b) I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
-
(a) Y. Liu, J. Shi, Y. Chen and C.-F. Ke, Angew. Chem., Int. Ed., 2008, 47, 7293–7296 CrossRef CAS PubMed;
(b) Y. Jiao, K. Liu, G. Wang, Y. Wang and X. Zhang, Chem. Sci., 2015, 6, 3975–3980 RSC;
(c) R. Joseph, A. Nkrumah, R. J. Clark and E. Masson, J. Am. Chem. Soc., 2014, 136, 6602–6607 CrossRef CAS PubMed;
(d) K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285–8289 CrossRef CAS PubMed.
-
(a) N. Basilio, M. Martín-Pastor and L. García-Río, Langmuir, 2012, 28, 6561–6568 CrossRef CAS PubMed;
(b) Y. Cao, X. Hu, Y. Li, X. Zou, S. Xiong, C. Lin, Y. Shen and L. Wang, J. Am. Chem. Soc., 2014, 136, 10762–10769 CrossRef CAS PubMed;
(c) K. Jie, Y. Zhou, Y. Yao, B. Shi and F. Huang, J. Am. Chem. Soc., 2015, 137, 10472–10475 CAS;
(d) Y. Chang, K. Yang, P. Wei, S. Huang, Y. Pei, W. Zhao and Z. Pei, Angew. Chem., Int. Ed., 2014, 53, 13126–13130 CrossRef CAS PubMed.
-
(a) K. M. Park, J. A. Yang, H. Jung, J. Yeom, J. S. Park, K. H. Park, A. S. Hoffman, S. K. Hahn and K. Kim, ACS Nano, 2012, 6, 2960–2968 CrossRef CAS PubMed;
(b) J. Zhao, C. Chen, D. Li, X. Liu, H. Wang, Q. Jin and J. Ji, Polym. Chem., 2014, 5, 1843–1847 RSC.
-
(a) W. L. Mock and N.-Y. Shih, J. Org. Chem., 1983, 48, 3618–3619 CrossRef CAS;
(b) W. L. Mock and N.-Y. Shih, J. Org. Chem., 1986, 51, 4440–4446 CrossRef CAS.
-
(a) G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489–19497 CrossRef CAS PubMed;
(b) R. Shi, Y. Chen, X. Hou and Y. Liu, RSC Adv., 2016, 6, 15175–15179 RSC;
(c) Q. Zhang, X. Yao, D.-H. Qu and X. Ma, Chem. Commun., 2014, 50, 1557–1567 Search PubMed.
-
(a) J. Wang, H. Y. Zhang, X. J. Zhang, Z. H. Song, X. J. Zhao and Y. Liu, Chem. Commun., 2015, 51, 7329–7332 RSC;
(b) I. Tomatsu, A. Hashidzume and A. Harada, Macromolecules, 2005, 38, 5223–5227 CrossRef CAS;
(c) I. Tomatsu, A. Hashidzume and A. Harada, Macromol. Rapid Commun., 2005, 26, 825–829 CrossRef CAS.
-
(a) A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS;
(b) T. M. Allen, C. Hansen, F. Martin, C. Redemann and A. Yau-Young, Biochim. Biophys. Acta, Biomembr., 1991, 1066, 29–36 CrossRef CAS;
(c) A. K. Dash and G. C. Cudworth, J. Pharmacol. Toxicol. Methods, 1998, 40, 1–12 CrossRef CAS PubMed.
-
(a) H. S. Yoo and T. G. Park, J. Controlled Release, 2001, 70, 63–70 CrossRef CAS;
(b) H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2014, 4, 81–89 CrossRef CAS PubMed.
- Y.-M. Zhang, Y. Yang, Y.-H. Zhang and Y. Liu, Sci. Rep., 2016, 6, 28848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21900d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.