
Determining chemospecificity in reactions with chain transfer agent and corresponding radical via evaluation of molecular weight dependency of apparent comonomer reactivity ratios: free-radical copolymerization of vinyl acetate and dibutyl maleate†
 *b
*b
Abstract
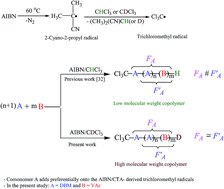
Free-radical copolymerization of vinyl acetate (VAc) and dibutyl maleate (DBM) in deuterated chloroform (CDCl3) initiated with 2,2′-azobis(isobutyronitrile) at 60 °C was monitored via online 1H-NMR kinetic experiment. The reactivity ratios of the VAc (rVAc) and DBM (rDBM) were calculated using data obtained from two online experiments. Two, i.e. classic and new, approaches were used to collect conversion and copolymer composition data. Results showed that for a copolymerization with a relatively large difference between the reactivity ratios (rVAc to rDBM ratio of about 5) the reactivity ratios can be calculated by data collected only from one online 1H-NMR experiment. In the case of copolymerization with the chloroform where copolymers with a number-average molecular weight (Mn) equal to or below 104 g mol−1 were synthesized, rVAc and rDBM were found to be 0.1135 and 0.0562 (r1r2 = 0.0064), respectively, [J. Polym. Res., 2014, 21, 582]. In the present work where copolymers with an Mn value of about 2 × 104 g mol−1 were synthesized, rVAc and rDBM were found to be 0.1911 and 0.0381 (r1r2 = 0.0073), respectively. The chemospecificity of the chloroform and corresponding trichloromethyl (CCl3) radical in the reaction with the propagating radical and comonomers, respectively, was deduced from instantaneous copolymer composition curves. The results revealed that DBM adds preferentially onto the CCl3 radicals derived from AIBN/CHCl3 or AIBN/CDCl3, indicating that copolymer composition and hence apparent reactivity ratios can be affected by the copolymer's molecular weight. Results may also be attributed to the preferential addition of the propagating radicals with a terminal DBM unit to CHCl3 or CDCl3.
 Please wait while we load your content...
Please wait while we load your content...

