
Converting urea into high value-added 2-oxazolidinones under solvent-free conditions†
Abstract
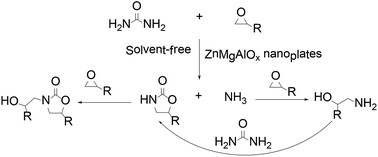
Zn-modified mesoporous Mg–Al nanoplates oxides were prepared by co-precipitation and further characterized and used in the synthesis of 2-oxazolidinones from urea and epoxides under solvent-free conditions. The characterization results suggested that Zn1.1Mg2.0AlO4.6, which featured more accessible active medium basic sites, were favorable for obtaining superior catalytic activity. This synthetic process is mild, convenient, simple and gives good yields up to 80%.
 Please wait while we load your content...
Please wait while we load your content...

