
Polymer–protein hybrid scaffolds as carriers for CORM-3: platforms for the delivery of carbon monoxide (CO)†
Diep Nguyena,
Susan Oliverab,
Nik Nik M. Adnana,
Cristan Herbertc and
Cyrille Boyer*ab
aCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, UNSW Australia, Sydney, Australia 2052. E-mail: cboyer@unsw.edu.au
bAustralian Centre for Nanomedicine (ACN), UNSW Australia, Sydney, Australia 2052
cSchool of Medical Sciences, UNSW Australia, Sydney, Australia 2052
First published on 22nd September 2016
Abstract
Recognised as a signalling molecule in mammals, carbon monoxide (CO) has a number of beneficial effects, including anti-inflammatory, anti-apoptotic, anti-proliferative and cytoprotective properties, and has been evaluated in clinical trials as a novel therapy. However, the clinical use of CO gas has been hampered due to safety issues and difficulties with delivery to specific sites of action. These challenges have led chemists to explore CO-releasing molecules (CORMs) as an alternative to administration of CO gas. [fac-RuCl(κ2-H2NCH2CO2)(CO)3] (CORM-3), (tricarbonylchloro(glycinato)ruthenium(II)), in particular, has gained increasing attention in biological studies due to its commercial water-soluble CORM and its remarkable biological effects. However, this compound possesses some unfavourable characteristics that have limited its clinical applications, including short CO-releasing half-life, poor stability and low cellular uptake efficiency. These issues can be prevented by incorporating CORM-3 into macromolecular scaffolds. In this study, we present for the first time the use of a polymer–protein hybrid system as a CORM-3 carrier. Firstly, the pyridyl disulfide-functional polymer (2-hydroxy ethyl acrylate) was prepared via Reversible Addition-Fragmentation chain-Transfer (RAFT) polymerisation. The obtained polymer was subsequently immobilised to protein, bovine serum albumin (BSA), via a “grafting to” approach. CORM-3 was then incorporated onto the protein–polymer conjugate BSA–P(HEA) via coordination interaction with histidine residues on BSA. A myoglobin assay demonstrated the CORM-3-functionalised polymer–protein conjugate released CO in a controlled manner in a buffer (pH 7.4) solution, providing slower release compared with CORM-3.
Introduction
Carbon monoxide has potential as a therapeutic agent owing to its beneficial effects in mammals, including anti-inflammatory, anti-apoptotic, anti-proliferative, anti-microbial and cytoprotective effects.1–8 However, the challenge of storing and delivering CO to the target tissue in a controlled and measurable fashion has hindered its clinical use because CO is a gaseous molecule and toxic at high concentrations. Thus, CORMs (CO releasing molecules)4,6 have been explored as a safer and more convenient system for delivering CO. Among the early-developed and extensively studied CORMs in biological systems is CORM-3 (tricarbonylchloro(glycinato)ruthenium(II)). This compound exerts similar effects to those observed for CO inhalation in a multitude of animal models of inflammatory disease,9–11 cardiovascular disease12,13 as well as organ transplantation and preservation.14 Clinical applications are limited, however, owing to major problems associated with this compound, including short CO-releasing half-life, poor stability in aqueous environments and low cellular uptake efficiency. These issues can be addressed by conjugating CORM-3 to macromolecular scaffolds.Proteins have gained increasing attention as CORM carriers owing to their unique characteristics, namely low toxicity, stability, biodegradability, non-immunogenicity and their ease of preparation.16–19 CORM-3 can be attached to protein via a coordination interaction with histidine (His) residues on the protein.15,20 His-15 in hen egg white lysozyme (HEWL) was demonstrated to react selectively with RuII(CO)2 fragments derived from CORM-3 in aqueous solutions (Scheme 1).15 Bernardes group employed protein bovine serum albumin (BSA) as a scaffold for CO delivery both in vitro and in vivo.21 The CO-releasing protein-based system was prepared by the reaction of the hydrolytic composition product of CORM-3 with histidine residues exposed on the surface of BSA in aqueous solution. Nondenaturating nanoelectrospray ionisation MS (native MS) demonstrated 7 out of 16 His residues per BSA molecule reacted with CORM-3.21 The prepared CO-releasing material supressed the production of pro-inflammatory cytokines, including interleukin (IL)-6, IL-10, and tumour necrosis factor (TNF)-a, by cancer cells.21
 | ||
| Scheme 1 Reaction of CORM-3 with the single His–protein HEWL in aqueous aerobic solution. Adapted from ref. 15. | ||
The CXC chemokine IL-8, which plays an important role in inflammatory response,22 was also used as a CO carrier.23 The CO-releasing moiety cis-[Ru(CO)2]2+ was reacted with selective His33 residues of IL-8 for 30 min in phosphate buffer solution (PBS) to yield IL-8-Ru(CO)2. The metalloprotein was found to release CO spontaneously in live HeLa cells and was non-toxic. Notably, neither the introduction of ruthenium carbonyl fragment nor the CO release from IL-8-Ru(CO)2 altered IL-8's neutrophil chemotactic activity. Therefore, protein shows promise for the safe and controlled delivery of CO into living cells or tissue.
Most protein-based drugs, however, exhibit rapid clearance and non-specific distribution of the drug, resulting in an inability to achieve sustained drug release.18,24–26 Incorporation of protein onto polymers to form protein–polymer conjugates can provide enhanced therapeutic effectiveness over the native protein, prolonging half-life of the protein in the blood stream, enhancing protein stability, decreasing immunogenicity and increasing the hydrodynamic diameter of the protein.27,28 Building upon the unique characteristics of protein–polymer conjugates, we aimed to develop a new CO delivery system using protein-based polymeric nanoparticles. To the best of our knowledge, no previous attempt has been made to explore polymer–protein hybrid scaffolds as platforms for CO delivery in biological systems.
In this article, we report for the first time the synthesis of protein-derived polymeric material for the delivery of CO. Firstly, pyridyl disulfide-functional polymer (2-hydroxy ethyl acrylate) (P(HEA)) was prepared via reversible addition–fragmentation chain-transfer (RAFT) polymerisation and subsequently reacted with BSA through a reversible disulfide bond. The successful conjugation of polymer to BSA was analysed via gel permeation chromatography (GPC) analysis and sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). CORM-3 was incorporated into the protein–polymer conjugate via coordination interaction to histidine residues on BSA. In comparison with free CORM-3, BSA–polymer-based CORM displayed slower CO release as observed by myoglobin assay.
Results and discussion
Preparation of pyridyl disulfide functional polymers P(HEA) and P(HEA) conjugated protein
To build a stable well-defined polymer–protein conjugate, we employed a “grafting to” approach involving a reactive group on the polymer coupling to a specific functional amino acid on the protein. Free thiols on cysteine residues29,30 are common targets for site specific conjugations. Several thiol-reactive groups have been used for modification of free thiols on proteins, including maleimides,31–34 vinyl sulfones,35,36 and thiol–disulfides.29,37–41 The pyridyl disulfide group is a particularly attractive functional group in bioconjugation procedures due to the fast formation of reversible disulfide linkages in thiol–disulfide exchange reactions. We have designed a polymer with a pyridyl dilsulfide group to conjugate with the cysteine residue on protein BSA (Cys-34) through a reversible disulfide bond. Utilising an Ellman's assay, we determined that 59 mol% of thiol residue was oxidised leaving 41 mol% of free thiol residue on BSA available for conjugation (Fig. S8, ESI†).Extensively used in drug delivery and bioconjugation,42,43 RAFT polymerisation is a robust method providing excellent control over molecular weight and the ability to introduce a variety of different moieties onto the polymer chain end. We chose a pyridyl disulfide functionalised RAFT agent for the polymerisation of 2-hydroxyethyl acrylate (HEA) affording a biocompatible and water soluble polymer44–49 with a pyridyl disulfide end group (Scheme 2). The polymerisation was conducted in DMF at 70 °C using the [HEA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[PDS-RAFT]:[AIBN] ratio of 200:1:0.1. The monomer conversion reached 90% after 2 h, which was calculated from 1H-NMR spectroscopy based on the integral ratio of the vinyl proton peak of monomer at 5.9 ppm to the ester signal at 4.2 ppm of crude reaction mixture. The resultant homopolymer P(HEA) had a number average molecular weight (Mn) of 23300 g mol−1 and a dispersity (Đ) of 1.20 as measured by gel permeation chromatography (GPC) analysis. The P(HEA) polymer structure was confirmed by 1H-NMR spectroscopy (Fig. 1) with characteristic signals at 4.2 and 3.8 ppm attributed to CH2O ester and CH2OH, respectively (as listed in Fig. 1) and pyridyl disulfide functional RAFT signals at 7.2–8.5 ppm attributed to phenyl protons on the pyridine group and the signal at 0.87–0.92 ppm corresponding to a methyl proton CH3 in the trithiocarbonate unit (–SCH2CH2CH2
:[PDS-RAFT]:[AIBN] ratio of 200:1:0.1. The monomer conversion reached 90% after 2 h, which was calculated from 1H-NMR spectroscopy based on the integral ratio of the vinyl proton peak of monomer at 5.9 ppm to the ester signal at 4.2 ppm of crude reaction mixture. The resultant homopolymer P(HEA) had a number average molecular weight (Mn) of 23300 g mol−1 and a dispersity (Đ) of 1.20 as measured by gel permeation chromatography (GPC) analysis. The P(HEA) polymer structure was confirmed by 1H-NMR spectroscopy (Fig. 1) with characteristic signals at 4.2 and 3.8 ppm attributed to CH2O ester and CH2OH, respectively (as listed in Fig. 1) and pyridyl disulfide functional RAFT signals at 7.2–8.5 ppm attributed to phenyl protons on the pyridine group and the signal at 0.87–0.92 ppm corresponding to a methyl proton CH3 in the trithiocarbonate unit (–SCH2CH2CH2![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3) (Fig. 1).
3) (Fig. 1).
 | ||
| Scheme 2 Schematic diagram for preparation of CORM-3 functionalised polymer–protein hybrid BSA–P(HEA)-CORM-3. | ||
 | ||
| Fig. 1 1H-NMR spectra of P(HEA) using PDS-RAFT as RAFT agent. | ||
The functionalised polymer P(HEA) was subsequently reacted with BSA – the conjugation reaction was carried out in PBS (pH 8.2) for 24 h employing a large excess of polymer ([BSA]:[P(HEA)] = 1:30) to guarantee complete conjugation. After dialysis against deionised (DI) water (MWCO = 50 K), the sample was analysed by GPC and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The retention time of the polymer–protein conjugate P(HEA)–BSA, measured by GPC, shifted to the left (Fig. S5, ESI†) compared with the native protein BSA, confirming the successful attachment of polymer to protein. Additionally, the polymer–protein conjugate was observed at higher molecular weight (Fig. 2, lane 3 and 4) than the native BSA (Fig. 2, lane 2) as shown by SDS-PAGE. The final product comprised a mixture of BSA–P(HEA) and unreacted BSA (as observed in Fig. 2, lane 3) owing to the fact that the commercial BSA contained a mixture of oxidised and non-oxidised cysteine residues.40,50 This was also confirmed by Ellman's assay, which showed that 41% of the Cys-34 on BSA was available for conjugation (Fig. S8, ESI†). After reaction with polymer, we did not observe free thiol, which suggests that one P(HEA) polymer was conjugated to BSA. In the presence of 2-mercaptoethanol (reducing conditions), the disulfide bond between the protein and polymer was cleaved, hence, the reduced BSA–P(HEA) (Fig. 2, lane 6) stayed at the same position compared to reduced BSA (Fig. 2, lane 5). Both aqueous GPC and SDS-PAGE indicated the successful conjugation of polymer with a pyridyl disulfide chain end to protein BSA through a reversible disulfide bond.
 | ||
| Fig. 2 SDS-PAGE visualised by Coomassie blue staining. Lane (1): protein marker, (2): non-reduced BSA; (3): non-reduced BSA–P(HEA); (4): non-reduced BSA–P(HEA)-CORM-3; (5): reduced BSA; (6): reduced BSA–P(HEA); (7): reduced BSA–P(HEA)-CORM-3. Note: the amount of protein in each sample is approximately 0.5 mg mL−1. | ||
Attachment of CORM-3 to polymer–protein conjugate BSA–P(HEA)
CORM-3 has been demonstrated to react with BSA via a coordination interaction with histidine (His) residues on the protein.15,20,21,23 Therefore, the polymer–protein hybrid BSA–P(HEA) can interact with CORM-3. The CO-releasing macromolecule was simply prepared by mixing BSA–P(HEA) with 50 equivalents of CORM-3 in phosphate buffer solution (PBS, pH 7.4) for 1 h. The mixture was then dialysed against DI water to remove unreacted CORM-3. After purification, the IR spectrum of the CORM-3 conjugated polymer–protein hybrid vehicle (Fig. 3) showed an additional characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band in the region from 1900–2100 cm−1 compared with the blank polymer–protein conjugate, confirming attachment of CORM-3 to the polymer–protein conjugate BSA–P(HEA). To determine the number of His residues on BSA–P(HEA) reacted with CORM-3, the ruthenium content was measured using Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES). ICP-OES analysis showed that Ru content in BSA–P(HEA)-CORM-3 was 0.69%, which corresponds to an average of 6 out of 16 His residue on BSA being modified and is consistent with the data reported by Bernardes and co-workers.21 In addition, successful conjugation of CORM-3 to the polymer–protein hybrid (Fig. 2) was demonstrated by SDS-PAGE. As expected, in both non-reducing conditions and reducing conditions, the CORM-3 functionalised polymer–protein hybrid BSA–P(HEA)-CORM-3 (Fig. 2, lane 4 and lane 7) appeared at a higher molecular weight peak compared with blank BSA–P(HEA) (Fig. 2, lane 3 and lane 6). It should be noted that a reaction between CORM-3 and a protein–polymer hybrid BSA–P(HEA) that contained a mixture of modified BSA–P(HEA) and unmodified BSA would result in a mixture of modified BSA–P(HEA)-CORM-3 and BSA-CORM-3.
O band in the region from 1900–2100 cm−1 compared with the blank polymer–protein conjugate, confirming attachment of CORM-3 to the polymer–protein conjugate BSA–P(HEA). To determine the number of His residues on BSA–P(HEA) reacted with CORM-3, the ruthenium content was measured using Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES). ICP-OES analysis showed that Ru content in BSA–P(HEA)-CORM-3 was 0.69%, which corresponds to an average of 6 out of 16 His residue on BSA being modified and is consistent with the data reported by Bernardes and co-workers.21 In addition, successful conjugation of CORM-3 to the polymer–protein hybrid (Fig. 2) was demonstrated by SDS-PAGE. As expected, in both non-reducing conditions and reducing conditions, the CORM-3 functionalised polymer–protein hybrid BSA–P(HEA)-CORM-3 (Fig. 2, lane 4 and lane 7) appeared at a higher molecular weight peak compared with blank BSA–P(HEA) (Fig. 2, lane 3 and lane 6). It should be noted that a reaction between CORM-3 and a protein–polymer hybrid BSA–P(HEA) that contained a mixture of modified BSA–P(HEA) and unmodified BSA would result in a mixture of modified BSA–P(HEA)-CORM-3 and BSA-CORM-3.
 | ||
| Fig. 3 ATR-FTIR spectra of polymer P(HEA), protein–polymer hybrid BSA–P(HEA) and CORM-3 conjugated protein–polymer hybrid BSA–P(HEA)-CORM-3. | ||
Myoglobin assay
In order to investigate whether polymer–protein conjugate BSA–P(HEA)-CORM-3 could be used as a novel macromolecular carrier of CORM-3 in vitro, the CO-release properties of polymer–protein based-CORM-3 were monitored spectrophotometrically by measuring the conversion of deoxymyoglobin (Deoxy-Mb) to carboxymyoglobin (MbCO). As Deoxy-Mb has strong affinity for CO, it can bind CO to generate MbCO.51 The spectral changes of reduced myoglobin solution with the addition of small aliquots of CO-releasing macromolecule BSA–P(HEA)-CORM-3 were recorded for an extended period of time at room temperature. Fig. 4 showed the changes in the UV-Vis spectra during the evolution from Deoxy-Mb to MbCO for 80 min. CORM-3 rapidly decomposes in aqueous solution and spontaneously releases CO with a half-life of only a few minutes in aqueous solution.51,52 The half-life of CORM-3 conjugated BSA was around 7.5 min (Fig. S7, ESI†), while CORM-3 conjugated polymer–protein hybrid liberated CO slowly with a half-life of approximately 30 min. In comparison with BSA-CORM-3 (Fig. S7, ESI†) the protein–polymer-based CORM displayed slower CO release, clearly demonstrating the advantage of our approach.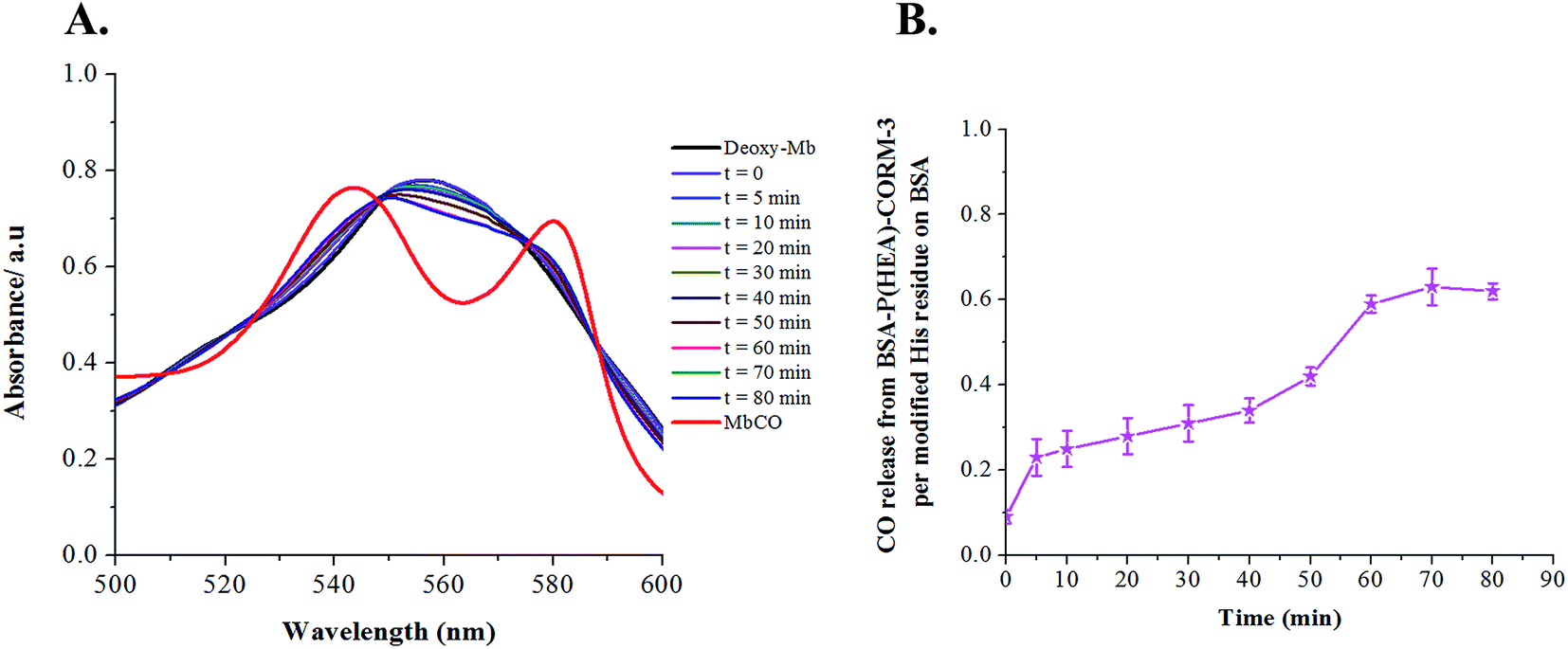 | ||
| Fig. 4 (A) UV-Vis spectra change of a solution of reduced horse skeletal muscle myoglobin in the presence of BSA–P(HEA)-CORM-3 (5 µM) in 0.1 M phosphate buffer solution (B) CO release from BSA–P(HEA)-CORM-3 (5 µM) versus time in reduced myoglobin solution. | ||
Conclusion
Our data describe a novel and simple approach for the delivery of CO utilising a polymer–protein material as the carrier. The CO-releasing polymer–protein material showed significant benefits over CORM-3 alone, including increased half-life of CORM-3. The half-life of CO from CORM-3 is very short (less than 1 min) while CORM-3 conjugated polymer–protein hybrid liberated CO slowly with a half-life of approximately 30 min. These features highlight the value of continued development of polymer–protein hybrid particles as macromolecular scaffolds for controlled delivery of CO.Acknowledgements
CB thanks the Australian Research Council (ARC) for his Future Fellowship (FT120100096).References
- X. Ji, K. Damera, Y. Zheng, B. Yu, L. E. Otterbein and B. Wang, J. Pharm. Sci., 2016, 105, 406–416 CrossRef CAS PubMed.
- F. Zobi, Future Med. Chem., 2013, 5, 175–188 CrossRef CAS PubMed.
- D. Nguyen and C. Boyer, ACS Biomater. Sci. Eng., 2015, 1, 895–913 CrossRef CAS.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs, 2005, 14, 1305–1318 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- S. Garcia-Gallego and G. J. Bernardes, Angew. Chem., Int. Ed. Engl., 2014, 53, 9712–9721 CrossRef CAS PubMed.
- D. Nguyen, N. N. M. Adnan, S. Oliver and C. Boyer, Macromol. Rapid Commun., 2016, 37, 739–744 CrossRef CAS PubMed.
- D. Nguyen, T.-K. Nguyen, S. A. Rice and C. Boyer, Biomacromolecules, 2015, 16, 2776–2786 CrossRef CAS PubMed.
- S. Mizuguchi, J. Stephen, R. Bihari, N. Markovic, S. Suehiro, A. Capretta, R. F. Potter and G. Cepinskas, Am. J. Physiol.: Heart Circ. Physiol., 2009, 297, H920–H929 CrossRef CAS PubMed.
- K. Tsoyi, T. Y. Lee, Y. S. Lee, H. J. Kim, H. G. Seo, J. H. Lee and K. C. Chang, Mol. Pharmacol., 2009, 76, 173–182 CrossRef CAS PubMed.
- S. Lancel, S. M. Hassoun, R. Favory, B. Decoster, R. Motterlini and R. Neviere, J. Pharmacol. Exp. Ther., 2009, 329, 641–648 CrossRef CAS PubMed.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, e2–e8 CrossRef CAS PubMed.
- Y. Guo, A. B. Stein, W. J. Wu, W. Tan, X. Zhu, Q. H. Li, B. Dawn, R. Motterlini and R. Bolli, Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H1649–H1653 CrossRef CAS PubMed.
- A. Bagul, S. A. Hosgood, M. Kaushik and M. L. Nicholson, Transplantation, 2008, 85, 576–581 CrossRef CAS PubMed.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 157, 168–182 CrossRef CAS PubMed.
- L. Chen, G. E. Remondetto and M. Subirade, Trends Food Sci. Technol., 2006, 17, 272–283 CrossRef CAS.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- S. Gunasekaran, S. Ko and L. Xiao, J. Food Eng., 2007, 83, 31–40 CrossRef CAS.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romao and M. J. Romao, Curr. Med. Chem., 2011, 18, 3361–3366 CrossRef CAS PubMed.
- M. Chaves-Ferreira, I. S. Albuquerque, D. Matak-Vinkovic, A. C. Coelho, S. M. Carvalho, L. M. Saraiva, C. C. Romão and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2015, 54, 1172–1175 CrossRef CAS PubMed.
- D. J. Brat, A. C. Bellail and E. G. Van Meir, Neuro-Oncology, 2005, 7, 122–133 CrossRef CAS PubMed.
- I. S. Albuquerque, H. F. Jeremias, M. Chaves-Ferreira, D. Matak-Vinkovic, O. Boutureira, C. C. Romao and G. J. L. Bernardes, Chem. Commun., 2015, 51, 3993–3996 RSC.
- L. R. Brown, Expert Opin. Drug Delivery, 2005, 2, 29–42 CrossRef CAS PubMed.
- V. P. Torchilin and A. N. Lukyanov, Drug Discovery Today, 2003, 8, 259–266 CrossRef CAS PubMed.
- B. J. Bruno, G. D. Miller and C. S. Lim, Ther. Delivery, 2013, 4, 1443–1467 CrossRef CAS PubMed.
- N. V. Katre, Adv. Drug Delivery Rev., 1993, 10, 91–114 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS PubMed.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- E. M. Pelegri-O'Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- Y. Jiang, M. Liang, D. Svejkar, G. Hart-Smith, H. Lu, W. Scarano and M. H. Stenzel, Chem. Commun., 2014, 50, 6394–6397 RSC.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS PubMed.
- E. Bays, L. Tao, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777–1781 CrossRef CAS PubMed.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS PubMed.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517–525 CrossRef CAS PubMed.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS PubMed.
- R. M. Broyer, G. N. Grover and H. D. Maynard, Chem. Commun., 2011, 47, 2212–2226 RSC.
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847–2851 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS PubMed.
- B. Yang, Y. Zhao, S. Wang, Y. Zhang, C. Fu, Y. Wei and L. Tao, Macromolecules, 2014, 47, 5607–5612 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- A. Gregory and M. H. Stenzel, Expert Opin. Drug Delivery, 2011, 8, 237–269 CrossRef CAS PubMed.
- Y. Chan, T. Wong, F. Byrne, M. Kavallaris and V. Bulmus, Biomacromolecules, 2008, 9, 1826–1836 CrossRef CAS PubMed.
- D. Popescu, R. Hoogenboom, H. Keul and M. Moeller, Polym. Chem., 2010, 1, 878–890 RSC.
- K. Bian and M. F. Cunningham, Macromolecules, 2005, 38, 695–701 CrossRef CAS.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2010, 43, 7041–7047 CrossRef CAS.
- O. V. Khutoryanskaya, Z. A. Mayeva, G. A. Mun and V. V. Khutoryanskiy, Biomacromolecules, 2008, 9, 3353–3361 CrossRef CAS PubMed.
- A. Vallés Lluch, A. Campillo Fernández, G. Gallego Ferrer and M. Monleón Pradas, J. Biomed. Mater. Res., Part B, 2009, 90, 182–194 Search PubMed.
- J. Janatova, J. K. Fuller and M. J. Hunter, J. Biol. Chem., 1968, 243, 3612–3622 CAS.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, E17–E24 CrossRef CAS PubMed.
- M. Desmard, R. Foresti, D. Morin, M. Dagouassat, A. Berdeaux, E. Denamur, S. H. Crook, B. E. Mann, D. Scapens, P. Montravers, J. Boczkowski and R. Motterlini, Antioxid. Redox Signaling, 2011, 16, 153–163 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterisation, Ellman's assay and UV spectroscopy spectra. See DOI: 10.1039/c6ra21703f |
| This journal is © The Royal Society of Chemistry 2016 |