
An oligothiophene chromophore with a macrocyclic side chain: synthesis, morphology, charge transport, and photovoltaic performance†
Swaminathan Venkatesana,
Jianyuan Suna,
Lianjie Zhang‡
a,
Ashish Dubeyb,
Andrew Sykesc,
Ting-Yu Lind,
Yu-Chueh Hungd,
Qiquan Qiaob and
Cheng Zhang*a
aDepartment of Chemistry and Biochemistry, South Dakota State University, SD 57007, USA. E-mail: cheng.zhang@sdstate.edu
bDepartment of Electrical Engineering, South Dakota State University, SD 57007, USA
cDepartment of Chemistry, University of South Dakota, Vermillion, SD 57069, USA
dInstitute of Photonics Technologies, National Tsing Hua University, Taiwan
First published on 19th October 2016
Abstract
An oligothiophene chromophore RingBDT(T3A)2 has been synthesized, where BDT is benzo[1,2-b:4,5-b′]dithiophene, Ring is a 1,12-dodecylenedioxy cyclic side chain on the benzene of BDT, T3 is 2,2′:5′,2′′-terthiophene, and A is an electron acceptor. In single crystals, the immediate precursor of RingBDT(T3A)2 formed π-dimers and the ring prevented further π-stacking of the dimers. A differential scanning calorimetry study showed that BDT(T3A)2, the ringless analog with two 2-ethylhexyloxy side chains on BDT, crystallized quickly from its melt upon cooling, while crystallization of RingBDT(T3A)2 melt upon cooling was slow and incomplete. Interestingly, RingBDT(T3A)2 solid crystallized fast at ∼110 °C upon heating, but its thin films (200 nm) remained amorphous after annealing at 80 °C. Despite the amorphous nature, the hole mobility of RingBDT(T3A)2 films (1.52 × 10−3 cm2 V−1 s−1) was 144% higher than that of the highly crystalline BDT(T3A)2 films (200–80 nm). Solar cells were fabricated from blends of the chromophores and phenyl-C61-butyric acid methyl ester (PC60BM). Thermal annealing at 100 °C for 10 minutes enhanced chromophore π–π interaction, and improved device fill factor and efficiency for the RingBDT(T3A)2 blend solar cells, while retaining the amorphous nature of blend. In stark contrast, thermal annealing under the same conditions caused the efficiency of BDT(T3A)2 cell efficiency to drop by 82%. This study demonstrates the effectiveness of using a macrocyclic side chain as a strategy for developing amorphous molecular semiconducting materials with improved mobility and morphological stability.
1. Introduction
Steady progress in bulk heterojunction (BHJ) solar cell device performance has been made in the past decade. Power conversion efficiencies of 10–11% have been achieved with fullerene derivative acceptors and conjugated polymer donors in the past two years.1–7 Solution-processed small molecule solar cells (SMSCs) have recently emerged as competitive alternatives to their polymer counterparts, exhibiting efficiencies as high as 10.1%.8–11 Regardless of the nature of donor materials, nanomorphology plays a crucial role in BHJs as it governs charge generation and charge transport,12,13 and is often tailored by optimization of the processing conditions such as solvent mixture,14 donor–acceptor mixing ratios, thermal annealing etc., as well as by structural modification of side chain15–17 and π-conjugated backbone.18–22Molecular chromophores with planar π-conjugated backbones tend to have strong crystallization tendency.23–30 Due to their small size, they can more easily form large crystals, making nanoscale bulk heterojunction difficult to obtain in BHJ devices. Low boiling point solvents such as chloroform are often used in spin coating of donor-phenyl-C61-butyric acid methyl ester (PC60BM) blend solutions to limit the size of donor crystalline domains.9,11,26–29,31–33 Even if nanomorphology can be achieved by using right solvent and processing additive,11 it can hardly be maintained upon thermal treatment or cycling. They require rather low optimal annealing temperatures (e.g., 75–80 °C (ref. 26 and 31)) as compared to those of polymer donors (e.g., 110 °C for poly(3-hexylthiophene) (P3HT)-PC60BM devices34). Annealing at moderately high temperatures (e.g., 120 °C) for a short time can cause a significant (28%) drop in performance,31 while annealing P3HT solar cells at 140 °C for 120 minutes causes little change in device efficiency.35
A common strategy to suppress crystallization is to add steric hindrance and conformational flexibility in molecular structures.36,37 For examples, bulky groups have been incorporated into oligothiophenes,38,39 polythiophene,40,41 perylene bisimide,42 poly(p-phenyleneethynylene)s,43 and oligo(p-phenylene-vinylene)s;44 cyclic side groups have also been used to provide steric protection for polythiophene,45 a thiophene-based fluorescent polymer, and porphyrin.46 Other strategies include blending structurally similar molecules, for examples, structurally similar acenes for thin film transistors,47 blends of PC61BM with PC71BM for BHJ solar cells,48,49 hole-transporting indolo[3,2-b]carbazole atropisomers50 and 8,8′-bis(indeno[2,1-b]thiophenylidene) atropisomers.51
In this paper, we report the design, synthesis and characterization of a oligothiophene chromophore with a macrocyclic side chain (ring) built on the benzo[1,2-b:4,5-b′]dithiophene (BDT) central unit. The new chromophore is named RingBDT(T3A)2 (Scheme 1), where T3 is 2,2′:5′,2′′-terthiophene, and A is an acceptor. In this design, the ring is to limit cofacial stacking to the formation of π-dimer, and the terthiophene segments are side chain-free to facilitate π–π interaction. With such design, we expect the chromophore to have low crystallization tendency, yet still have strong interchromophore interactions to facilitate charge transport. The ringless analog BDT(T3A)2 (Scheme 1) was also synthesized for comparative studies.
 | ||
| Scheme 1 Synthesis of chromophores RingBDT(T3A)2 and BDT(T3A)2. | ||
Single crystal XRD analysis of the immediate precursor of RingBDT(T3A)2 (compound 8) was performed to show how the ring affects molecular packing. Comparative studies were performed using proton nuclear resonance spectroscopy (1H NMR), differential scanning calorimetry (DSC), wide-angle X-ray diffraction, UV-vis absorption, time-of-flight and space charge limited current (SCLC) charge carrier mobility measurements. Effect of thermal annealing of chromophore films was investigated to find correlation between morphological stability and chromophore structure. Solar cell devices were fabricated from blends of chromophores and PC60BM. Processing additive and thermal annealing were used to improve device performance. Blend film morphology and charge mobility of the devices were studied to better understand the device performance.
2. Results and discussion
2.1 Chromophore synthesis
The synthesis of RingBDT(T3A)2 started from a ring-protected dibromobenzene (1).52 The key step in the reaction sequence was cyclization of compound 4 to form 5, a ring-protected BDT, which has not been reported. The reaction was performed in a diluted solution to minimize the intermolecular reaction between 4 and the product 5 which is more electron rich and thus more reactive toward aromatic electrophilic substitution than the benzene moiety in 4. The yield of 5 was 16% for three steps from 1. The dialdehyde 8 was obtained in a yield of 81% for two steps from 6. Knovenagel condensation of 8 and hexyl cyanoacetate led to RingBDT(T3A)2 in 79% yield (10% overall). This chromophore and all its precursors are like cyclophane derivatives and have planar chirality.53 So, RingBDT(T3A)2 and all intermediates were obtained as racemic mixtures. The structures shown in Scheme 1 are in the (S)-configuration.53The synthesis of the ringless analog BDT(T3A)2 is also shown in Scheme 1. The structure is only different from the literature chromophore10 in that it has hexyl groups, instead of octyl, at the two ends. Proton NMR spectra of 4, 5, 6, 8, RingBDT(T3A)2, and BDT(T3A)2, and 13C NMR spectra of 5, 6, RingBDT(T3A)2 are in Fig. S1–S10.†
2.2 Crystal structure of compound 8
Among 8, 10, RingBDT(T3A)2 and BDT(T3A)2, 8 has the lowest solubility (e.g., ∼4 mg mL−1 in chloroform). Both RingBDT(T3A)2 and BDT(T3A)2 are very soluble (>50 mg mL−1 in chloroform), even though RingBDT(T3A)2 contains much less side chains.Crystal growth was conducted for all chromophores in different solvents: chlorobenzene, mixture of chlorobenzene and diethyl ether or pentane, using different methods including slow cooling of saturated solution and dilution by poor solvent via vapor diffusion. Single crystals of sizes up to ∼2 × 2 × 1 mm were obtained for 8 from its heated chlorobenzene solution upon natural cooling. Transparent needle crystals of RingBDT(T3A)2 were obtained from its chlorobenzene solution, but were too small (∼0.02 mm) for single crystal XRD analysis. Under all crystallization conditions, 10 and BDT(T3A)2 only gave fine particles.
The crystals of 8 belong to the monoclinic space group P21/c with 4 molecules in the unit cell (Fig. S11†). All thiophene units are in the S-trans conformation, and the two aldehyde groups are S-cis to the sulfur atoms of the terminal thiophenes. The enantiomers pair up into a face-on-face dimer or π-dimer (Fig. 1a). The least-squares mean planes of two π systems have an average separation distance of 3.512 Å. The π–π stacking is tightest at the thiophene units that are next to the BDT with a separation of 3.231 Å (Fig. S12†), similar to that of a tetrathiophene end-capped with perfluorophenyl groups (∼3.20 Å),54 second tight at the center of the π-system with a distance of 3.745 Å and lest tight at the ends with a distance of 4.019 Å. The two molecules in the dimer pack with a slip of 1.539 Å along the long axis of the π system (Fig. 1b) and a slip of 0.405 Å along the short axis (Fig. 1c). Similar slippages have been reported for 5,6,11,12-tetrachlorotetracene single crystal.55
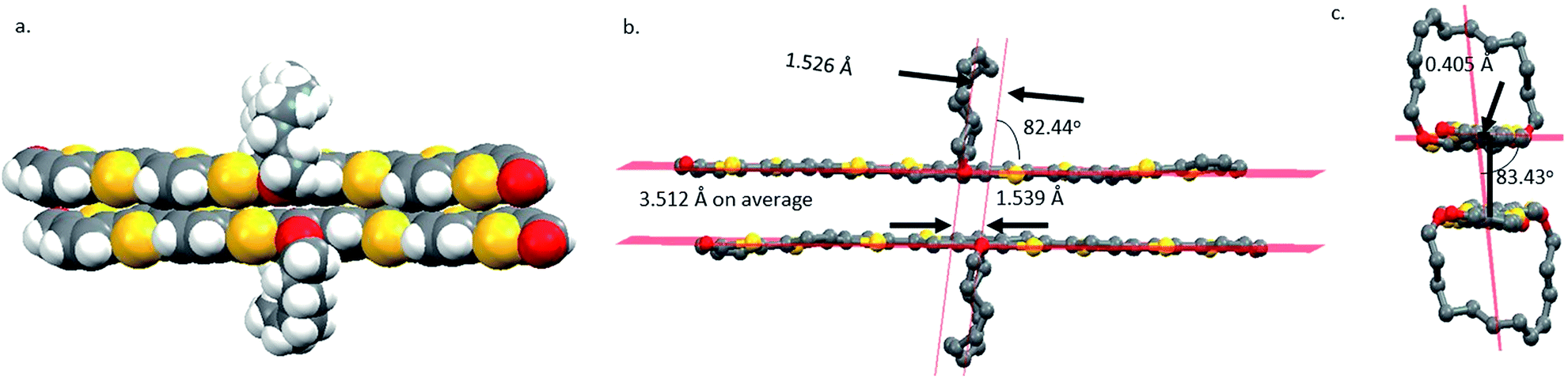 | ||
| Fig. 1 (a) The space-filling structure of 8 dimer in the crystal, (b) the ball-stick structure of the dimer, and (c) an end view of the dimer. | ||
Face-on-face stacking of 8 could not continue beyond the dimer due to ring protection for both sides. Instead, the dimers do slipped edge-on-edge stacking to form a one-dimensional (1D) array along the [1 0 1] direction (Fig. 2a). The average planes of all the dimers in an array are parallel, but have an offset of 0.500 Å between adjacent dimers (Fig. 2b). Two arrays of dimers stack co-linearly with one sitting with its side on the π plane of the other as shown in Fig. 2b and c (side and top views) and in Fig. S13† with more details. Clearly, there is no face-on-face stacking among all neighboring dimers. The repeated stacking of the 1D dimer arrays forms the crystal. When viewed in the [1 0 1] direction (Fig. 2d), the crystal is a bundle of an infinite number of 1D dimer arrays.
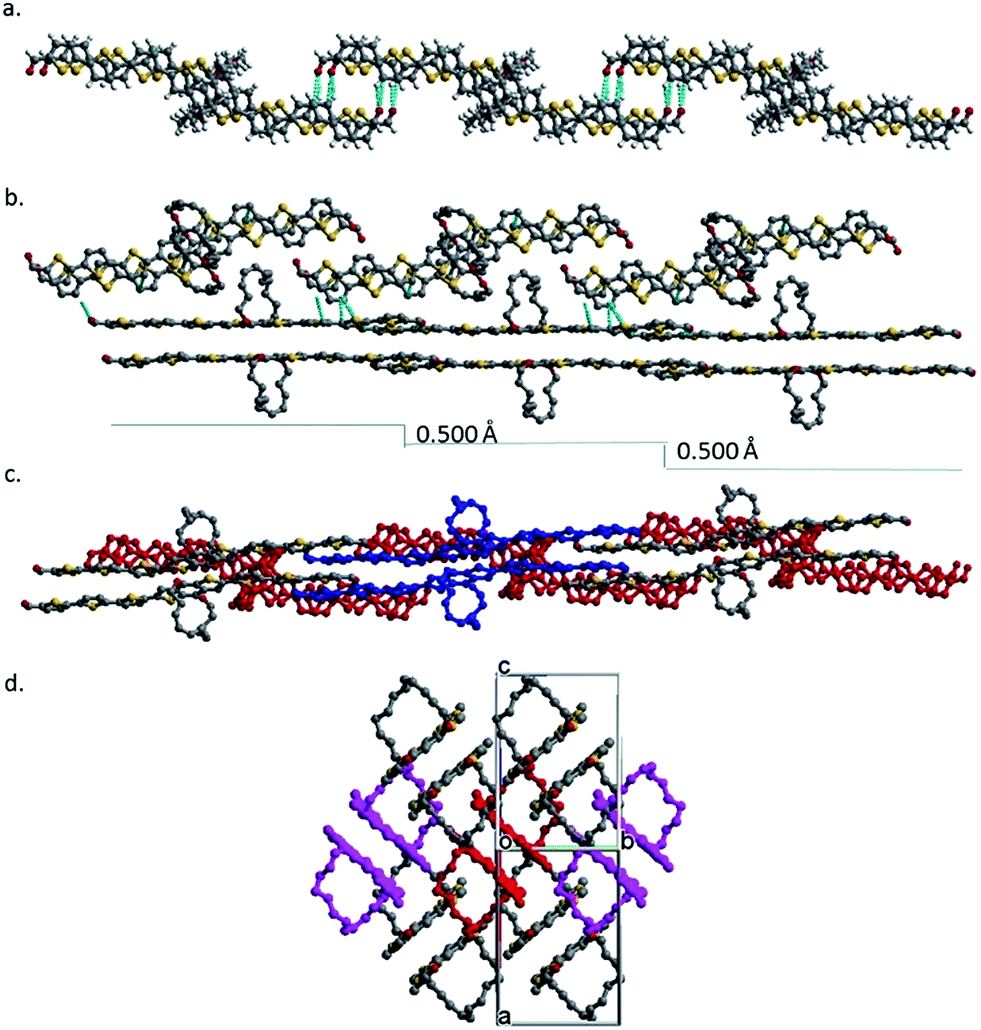 | ||
| Fig. 2 (a) A 1D 8 dimer array formed by slipped side-on-side stacking. (b) A stack of two 1D dimer arrays viewed from the side of the bottom array. (c) A stack of two 1D dimer arrays viewed from to top of the bottom array (colored in red). (d) An end view of a bundle of seven 1D arrays. | ||
2.3 1H NMR study of aggregation in solution
Würthner et al. have shown that a cyclic side chain can prevent perylene bisimides from aggregating beyond dimer in solution.56,57 To find the difference in aggregation behaviors of RingBDT(T3A)2 and BDT(T3A)2 in solution, proton NMR spectra of CDCl3 solutions of a series of concentration were obtained and are shown in Fig. 3. As the concentration was increased from 2.0 mM to 75 mM, shift of peaks to lower frequency was observed for all protons in the aromatic region, and, to a less extent, in the aliphatic region (Fig. S8†). For all concentrations, only one signal is observed for one proton. This can be perceived in terms of rapid exchange between monomers and aggregates on the NMR timescale. Chemical shifts of some protons (labeled in Scheme 1) are given in Fig. 3. The changes in chemical shift with increasing concentration are plotted in Fig. 3 right for protons a, g and h of RingBDT(T3A)2 and the corresponding protons in BDT(T3A)2 (a′, e′ and f′).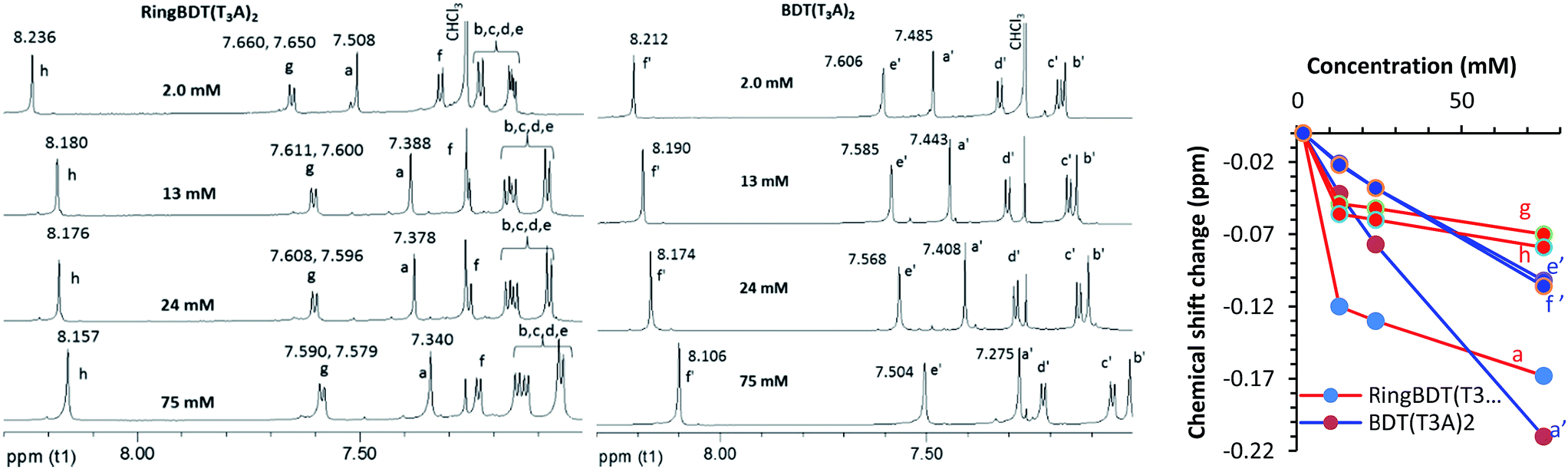 | ||
| Fig. 3 Proton NMR spectra of RingBDT(T3A)2 (left) and BDT(T3A)2 (middle), and 1H NMR chemical shift changes of selected protons of RingBDT(T3A)2 and BDT(T3A)2 with varying concentration (right). Solvent: CDCl3. Chemical shift changes are calculated with reference to the chemical shifts of each proton at 2.0 mM. | ||
First, proton a experiences greater change than g and h. This indicates that the π-systems have closer contact at the center than at the ends as found for 8 in the crystal; the same is observed for a′ vs. e′ and f′. Second, a, g and h curves underwent a sudden drop as the concentration was increased from 2.0 mM to 13 mM, and then decreased much slower from 13 mM to 75 mM, while a′, e′, and f′ curves decreased almost linearly over the whole range of concentration. The behavior of RingBDT(T3A)2 can be explained by formation of discrete dimers in the solutions, which was largely completed at 13 mM. Further increase in concentration continued to shift the equilibrium toward the dimer, but π-stacking of the dimers (i.e., formation of π-oligomers) did not happen as it would cause faster change in chemical shifts of g and h than that of a since the BDT units are sterically shielded by the rings and hence is less affected by π-stacking of dimers. Although chemical shift changes of BDT(T3A)2 are smaller than RingBDT(T3A)2 at low concentrations due to weaker π-interactions, they became larger at 75 mM due to formation of π-oligomers. Therefore, RingBDT(T3A)2 is self-limiting in π-stacking in solution. The similar observation was made in UV-vis absorption study of chromophore solutions of a wider range of concentrations (Section 2.6).
2.4 Thermal analysis of chromophore solids
Cyclic DSC analysis was performed for RingBDT(T3A)2 and BDT(T3A)2 particles which were precipitated from their chloroform solutions upon addition of EtOAc. A heating rate of 10 °C per minute and a cooling rate of 5 °C per minute were used. The DSC thermograms are shown in Fig. 4. BDT(T3A)2 showed a sharp melting peak at 214.2 °C upon each heating and a sharp crystallization peak at 190.1 °C upon cooling with a similar enthalpy change, indicating that crystallization of BDT(T3A)2 from melt is fast and complete.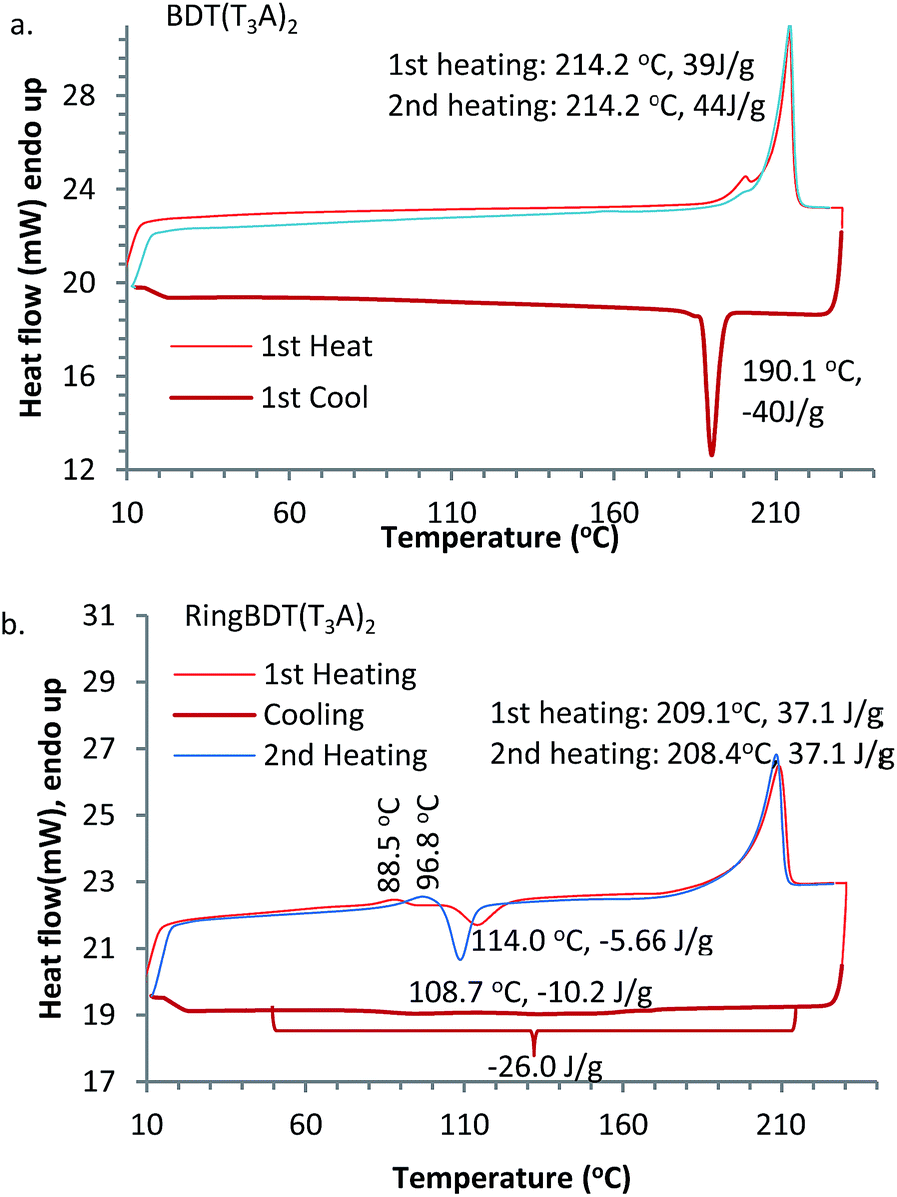 | ||
| Fig. 4 DSC thermograms of RingBDT(T3A)2 (a) and BDT(T3A)2 (b). Mass of sample: 6.2 mg. N2 flow rate: 20 mL min−1. Heating rate: 10 °C min−1. Cooling rate: 5 °C min−1. | ||
On the other hand, RingBDT(T3A)2 showed a slow crystallization process spanning from 210 °C to 50 °C in the cooling scan. A crystallization peak at ∼110 °C appeared in the first and the second heatings, indicating that the pristine solid sample was not fully crystalline and crystallization from the melt upon cooling was incomplete. It also showed a weak endothermic peak at 88.5 °C in the first heating, and 99 °C in the second heating, which is attributed to activation of thermal motion of side chains in amorphous domains. The shift of peak was likely caused by sample densification by the heating. The enthalpy changes of the melting peak at 208–209 °C are same for the two heating scans.
The DSC experiment was repeated with a fresh RingBDT(T3A)2 sample for four cycles (Fig. S14a†). The exothermic peak in the heating scan grew stronger and shifted from 114.3 °C to 108.7 °C from the first heating to the second, and became stabilized in the fourth heating. Similarly, among the cooling scans, the biggest change in the exotherm profile was between the first and second cooling scans, which are shown with magnification in Fig. S14b.† Since the highest temperature in the scans was far below the decomposition temperature, the difference was due to a change in physical form, presumably particle size. With more thermal cycling, the size became stabilized. The used sample was tested again for four cycles at a higher heating/cooling rate (20 °C per minute). The results are shown in Fig. S15.† From the first cooling on, the thermograms overlap completely, indicating that the particle size was stabilized. The endotherm of the melting process around 209 °C are practically same for all 8 cycles in the two experiments, suggesting that crystallization on heating is complete regardless of heating rate (5 or 20 °C min−1) and the sample history (either crystalline precipitates from solution or solid from cooling at 5 °C min−1 or 20 °C min−1). In Fig. S15,† the crystallization peak on the second heating (i.e., after the first fast cooling) is 36% stronger than that on the first heating, indicating that crystallization upon faster cooling was further reduced.
One-way scan from 10 °C to 400 °C was also performed for RingBDT(T3A)2, BDT(T3A)2, 8 and 10 (Fig. S16†). Ringless chromophores are more thermally stable, likely due to steric hindrance of the side chains to interchromophore reactions.
2.5 XRD and AFM study of RingBDT(T3A)2 and BDT(T3A)2 films
First, two solvents, chlorobenzene (CB, bp 131 °C) and chloroform (CF, bp 61 °C) were tested to find, with visual inspection, which produces films that are less scattering. It was found that BDT(T3A)2 films casted from a CB solution did not even show the color of chromophore, indicating the formation of micron-sized crystallites all over the surface. When CF was used, the films looked better in color, but were still more scattering than the RingBDT(T3A)2 films coated from its CB solution (see optical images in Fig. S18†). CF is also a good solvent for RingBDT(T3A)2. So, CF was chosen as the solvent for film preparation for both chromophores.X-ray diffractograms (Fig. 5a and b) were obtained for BDT(T3A)2 and RingBDT(T3A)2 thin films (thickness = 250 nm and 200 nm, respectively) spin-coated from their chloroform solutions on glass substrates. The BDT(T3A)2 thin film showed a strong (1 0 0) peak at 2θ = 4.51° and a (2 0 0) peak at 9.00°, corresponding to a d-spacing of 19.6 Å. The d-spacing is similar to that of P3HT (17.7 Å),58 suggesting a similar lamellar structure where π-stacks are separated by the hexyl side chains. In sharp contrast, no peaks were observed for RingBDT(T3A)2 in pristine films or films annealed at 80 °C for 10 minutes (Fig. 5b). A tiny peak showed up at 2θ = 5.70° only after annealing at 100 °C or 125 °C for 10 minutes. Surprisingly, two thick films (2.5–3.5 μm) annealed under the same conditions exhibited two major peaks at 5.75° and 24.45° (Fig. 5c), corresponding to d-spacings of 15.36 Å and 3.64 Å, respectively. Even an as-cast thick film showed weak signals near these two angles. The results indicate that RingBDT(T3A)2 crystallization is difficult in thin films. For comparison, the powder X-ray diffractogram of RingBDT(T3A)2 is shown in Fig. S17.† The strongest peak is at 2θ = 5.86°, accompanied by the second and third order peaks at 11.58° and 16.80°. There is no significant peak near 24.45°.
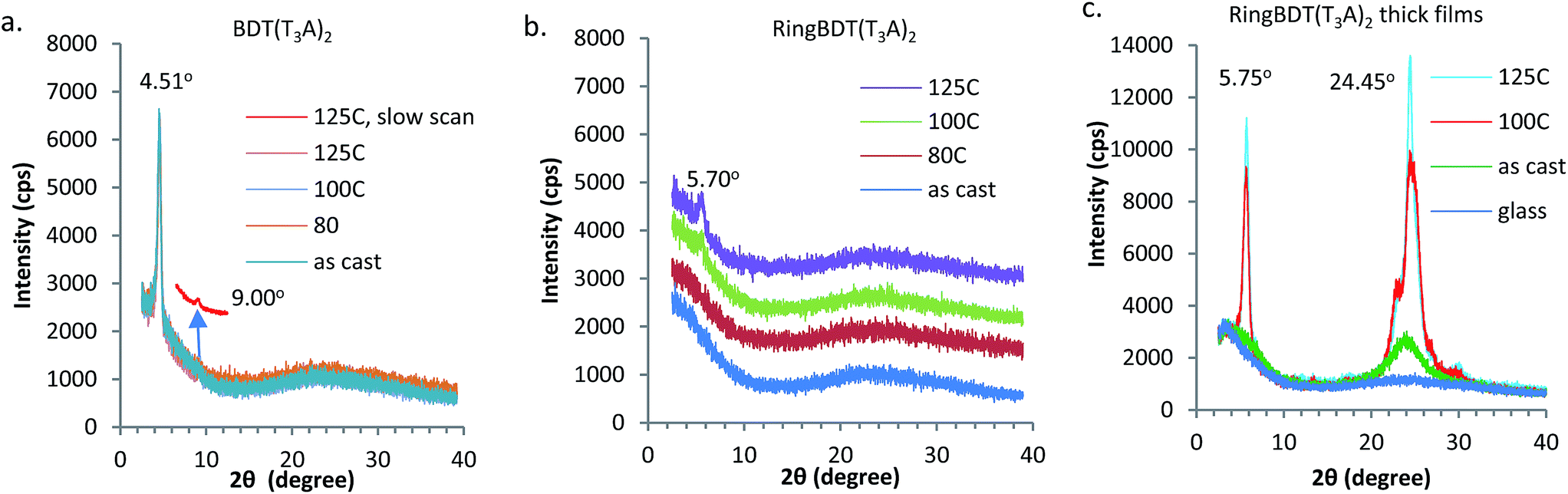 | ||
| Fig. 5 Wide angle X-ray diffractograms of (a) BDT(T3A)2 (250 nm) and (b) RingBDT(T3A)2 (200 nm) thin films, and (c) RingBDT(T3A)2 thick films (2.5–3.5 μm), as cast or annealed at different temperatures. The slow scan in (a) was performed at a scan rate of 0.25° min−1 to increase signal/noise ratio by 4 times. The thin films were spin coated from CHCl3 solutions. The thick films were drop casted from a chloroform solution. Scan speed: 4° min−1 and integration step: 0.01°. | ||
AFM topographic images were taken for films spin-coated on glass substrates from their chloroform solutions (Fig. 6). The BDT(T3A)2 films exhibits higher roughness (RMS roughness = 4.02 nm) and larger domains. On the other hand, RingBDT(T3A)2 films showed a smoother surface with RMS roughness = 0.58 nm.
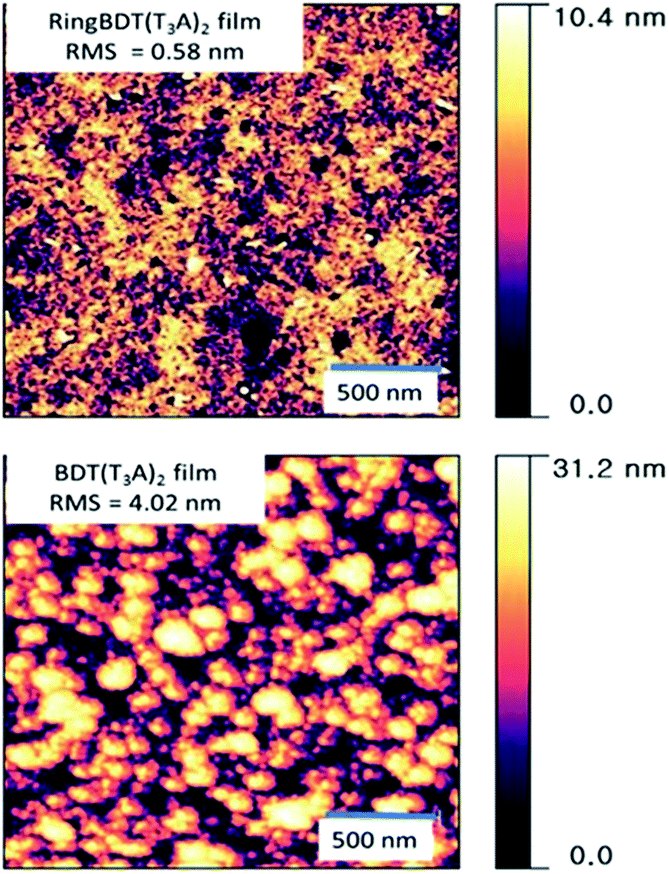 | ||
| Fig. 6 AFM surface topography (2 μm × 2 μm) of RingBDT(T3A)2 and BDT(T3A)2 thin films spin-coated from their chloroform solutions. Film thickness: ∼100 nm. | ||
2.6 UV-vis absorption spectra of solutions and films, and effect of annealing
Fig. 7 shows the optical absorption spectra of 0.01 mM chloroform solutions of RingBDT(T3A)2 and BDT(T3A)2. RingBDT(T3A)2 shows a longer peak wavelength (λmax) than BDT(T3A)2 (515 nm vs. 491 nm) (Fig. 7a). The peak extinction coefficient of RingBDT(T3A)2 solution (103![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 472 cm−1 M−1) is also higher than that of the BDT(T3A)2 solution (88459 cm−1 M−1). Both can be attributed to the better planarity of RingBDT(T3A)2 π-system due to the absence of alkyl groups on the thiophene units.
472 cm−1 M−1) is also higher than that of the BDT(T3A)2 solution (88459 cm−1 M−1). Both can be attributed to the better planarity of RingBDT(T3A)2 π-system due to the absence of alkyl groups on the thiophene units.
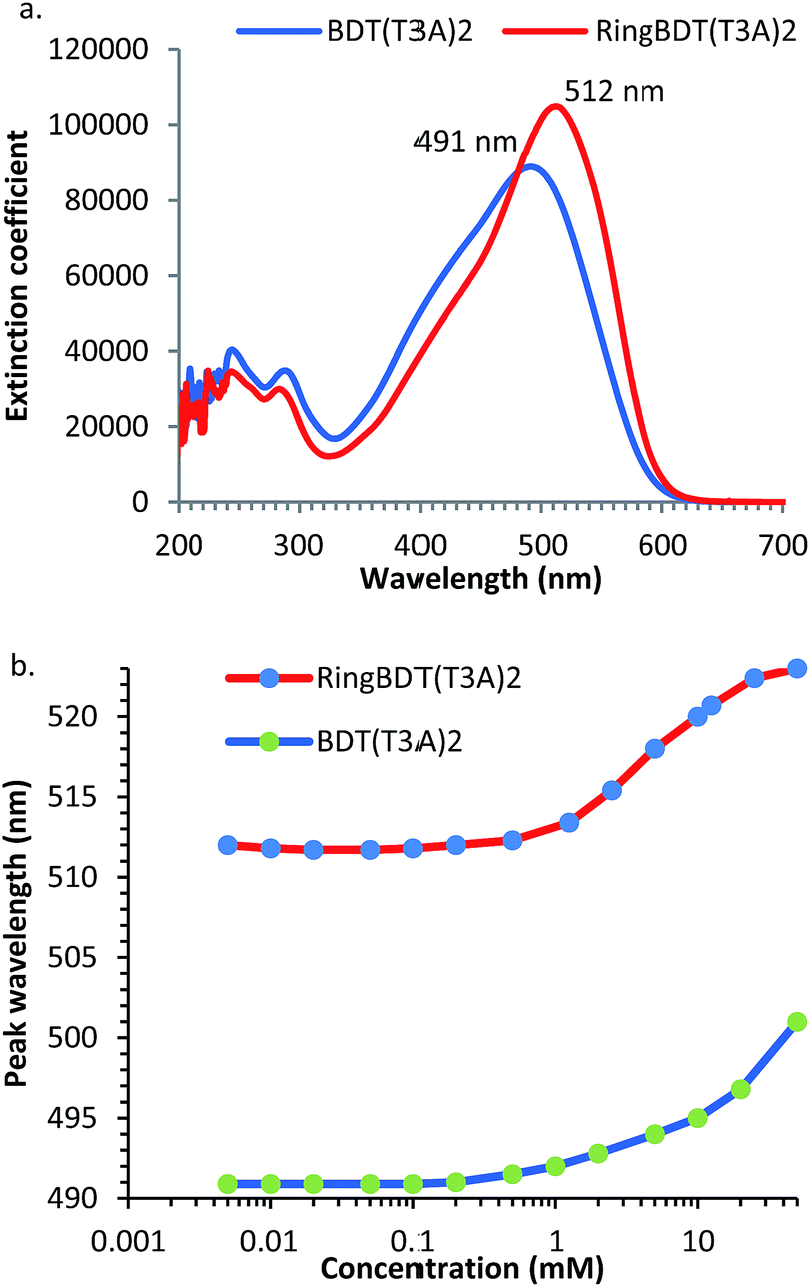 | ||
| Fig. 7 (a) UV-vis absorption spectra of 0.01 mM chloroform solutions of RingBDT(T3A)2 and BDT(T3A)2. (b) UV-vis absorption peak wavelength vs. concentration of chloroform solutions of RingBDT(T3A)2 and BDT(T3A)2. | ||
UV-vis absorption can also be used to study chromophore aggregation in solution. It complements the NMR method as it more conveniently measures solutions of very low concentrations. The spectra were recorded for both chromophores with concentrations ranging from 0.005 mM to 50 mM in chloroform, and are shown in Fig. S19 and S20.† λmaxs are plotted as a function of concentration in Fig. 7b λmax values of RingBDT(T3A)2 solutions increases very little from 0.005 mM to 0.5 mM, rise fast from 0.5 mM to 25 mM, and show sign of saturation from 10 mM to 50 mM. The sign of saturation is an indication that π-stacking of RingBDT(T3A)2 is limited to dimer formation in chloroform solution.
On the other hand, λmax of BDT(T3A)2 solution does not show a sign of saturation at 50 mM. The different behaviors are similar to those observed for 1H NMR chemical shift change (Section 2.2) of the two chromophores.
UV-vis absorption spectra of RingBDT(T3A)2 and BDT(T3A)2 films before and after thermal annealing are shown in Fig. 8. The films were casted from chloroform solutions on glass substrates. One film of each chromophore was annealed consecutively at 80 °C, 100 °C, and 125 °C, each for 10 minutes. The as-cast RingBDT(T3A)2 film (200 nm thick) had a λmax of 529 nm, a red shift of 17 nm from that of its dilute solution (0.01 mM), while BDT(T3A)2 exhibited a red shift of 60 nm. The larger red shift of BDT(T3A)2 is due to the non-planar molecular structure in solution and the crystalline packing in film.
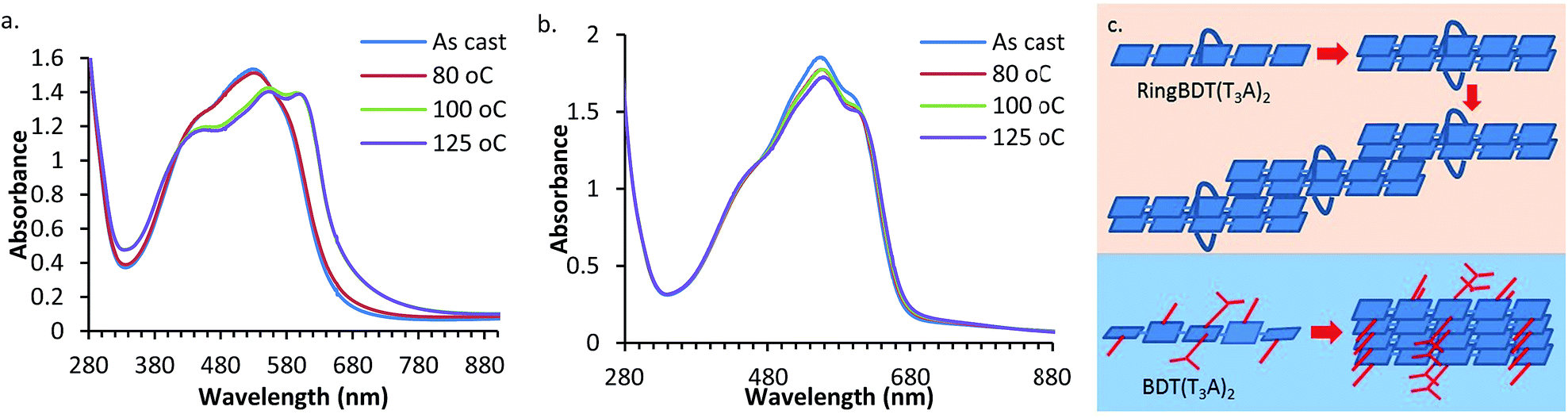 | ||
| Fig. 8 UV-vis absorption spectra of RingBDT(T3A)2 (a) and BDT(T3A)2 (b) films annealed at different temperatures for 10 minutes. Thicknesses are 200 nm and 275 nm, respectively. (c) Illustration of possible π-stacking structures of RingBDT(T3A)2 and BDT(T3A)2. | ||
For RingBDT(T3A)2, annealing at 80 °C for 10 minutes did not produce a notable change in the spectral profile; annealing at 100 °C for 10 minutes broadened the full width at half maximum (FWHM) by ∼25 nm, sharpened the vibronic features, and lowered the optical bandgap from 1.90 eV for as-cast film to 1.84 eV. Since XRD study of RingBDT(T3A)2 films of the same thickness did not show significant diffraction peaks after annealing at 100 °C and 125 °C (Fig. 5b), the stronger π-interaction exhibited in the film annealed at these temperatures is likely an indication that a staircase structure as illustrated in Fig. 8c was formed from half-side face-on-face π-stacking of the dimers. Such structure is amorphous, and appears to be quite stable since further annealing at 125 °C for 10 minutes did not produce any further change in the spectrum.
On the other hand, BDT(T3A)2 film (275 nm thick) exhibited no significant change in spectral profile upon annealing at 80 °C, 100 °C and 125 °C, and the optical bandgap only experienced a slight change from 1.85 eV before annealing to 1.84 eV after annealing at 125 °C. The result of this study supports the conclusion from the DSC analysis that BDT(T3A)2 crystallizes much faster than BDT(T3A)2 and crystallization is nearly complete upon film formation. Fast crystallization is expected for BDT(T3A)2 as it can form regular 1D π-stacks illustrated in Fig. 8c. The data of UV-vis absorption of 0.01 mM solutions and as-cast and 125 °C-annealed films are summarized in Table 1.
| λmax/nm, CHCl3a/film/125 °C | λcutoff/nm, CHCl3/film/125 °C | EOptg film (eV), as cast/125 °C | Ered/oxb (V) | LUMO/HOMO (eV) | EECg (eV) | |
|---|---|---|---|---|---|---|
| a 0.01 mM chloroform solutions.b Referenced to ferrocene oxidation onset potential which is 0.00 V against a Ag/AgNO3 reference electrode. | ||||||
| RingBDT(T3A)2 | 512/529/553 | 594/653/673 | 1.90/1.84 | −1.28/0.57 | −3.82/−5.67 | 1.85 |
| BDT(T3A)2 | 491/551/558 | 586/670/675 | 1.85/1.84 | −1.34/0.46 | −3.76/−5.56 | 1.80 |
2.7 Electrochemical and electrical characterization
Cyclic voltammograms of RingBDT(T3A)2 and BDT(T3A)2 are shown in Fig. 9a and b. In both plots, the signals of the negative scans are very weak and are scaled up by 6 times for a better view. The weak signals were not from reduction of dissolved oxygen as was confirmed by a blank scan (Fig. 9a). Weak reduction current was not reported for the solution CV of the BDT(T3A)2 analog,10 or any other polymers or oligomers, to our knowledge. However, this have been observed in film CVs of oligomeric chromophores59–62 and polymers.63–65 For films of some polymers64,66 and oligomeric chromophores,30,67–71 the reduction currents are even hard to measure. Slow diffusion of electrolyte ion was found responsible for weak CV current of some polymers.72,73 It is evident from the following observations that it is also the reason behind the low reduction currents of RingBDT(T3A)2 and BDT(T3A)2.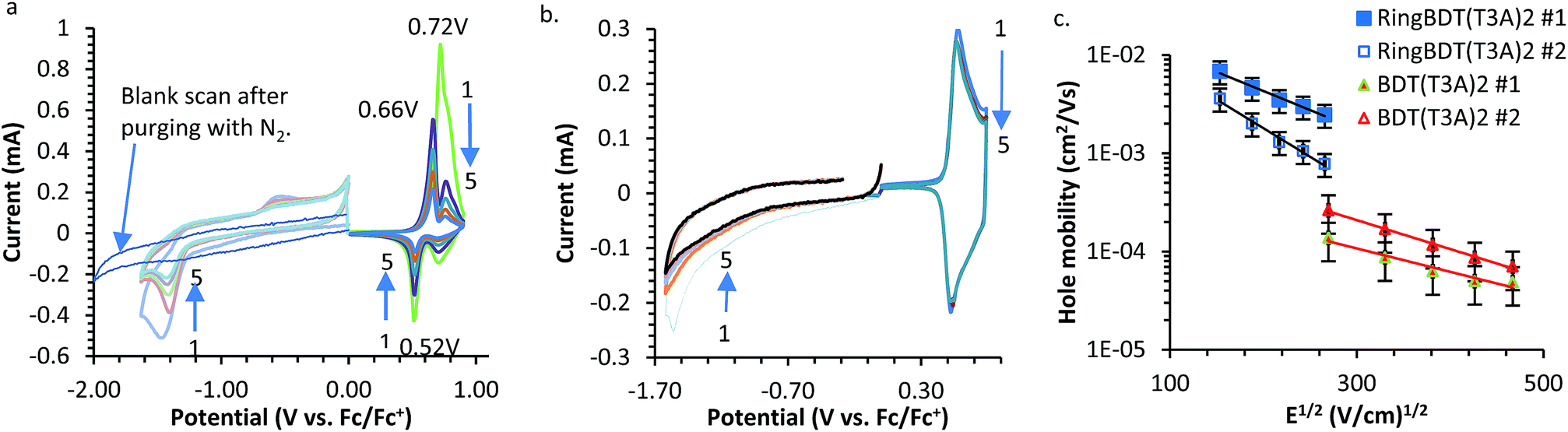 | ||
| Fig. 9 Cyclic voltammograms of RingBDT(T3A)2 (a) and BDT(T3A)2 (b). The currents of the negative scans (including the blank scan) are very small, and are scaled up by 6 times for a better view. Reference electrode: Ag/AgNO3. (c) Time of flight mobility measurements. Arrows in (a) and (b) indicates the trends of current from the first cycle to the last cycle. | ||
• The reduction current is much smaller than the oxidation current. The counter ion needed for reduction is tetrabutylammonium ion (TBA), which is much larger than PF6−.
• Both the reduction and oxidation currents of BDT(T3A)2 are 2–3 times greater than those of RingBDT(T3A)2 (Fig. 9a and b). This is because BDT(T3A)2 is highly crystalline, hard for counter ions to diffuse through.
• Thick films gave much lower oxidation and reduction currents (Fig. S22†).
In the positive scans of RingBDT(T3A)2, the intensity of the initial oxidation peak at 0.72 V quickly dropped, accompanied by appearance of a new reduction peak at 0.52 V and a new oxidation peak at 0.66 V. Solution UV-vis absorption spectrum of chromophore coating on Pt wire (scanned to 0.9 V) indicated that the chromophore was not decomposed upon the first oxidation. Presumably, the new peaks resulted from electrochemical conditioning of the film (i.e. annealing). The gradual decrease of all peaks was due to gradual dissolution of RingBDT(T3A)2 upon oxidation. The red/ox waves in the negative scans were not reversible at all. The chromophore was not dissolved during the negative potential cycling.
Oxidation of BDT(T3A)2 was reversible over five cycles. Its reduction was not reversible. Wider range CV scans were also performed for both chromophores (Fig. S21†) to show more oxidation and reduction states.
The redox potentials and electrochemical energy gaps of the chromophores are listed in Table 1. The HOMO/LUMO energies were calculated from the onset of oxidation/reduction potential relative to the onset oxidation potential of ferrocene (Eox/Ered) according to the following equation, HOMO or LUMO = −[5.1 + (Eox or Ered)] eV, where −5.1 eV is the HOMO energy of ferrocene updated by Guillermo Bazan in 2011.74 Solution CV was also performed for the chromophores in dichloromethane (Fig. S23†). The oxidations are partially reversible. The reductions are totally irreversible. The redox potentials of BDT(T3A)2 are nearly same as those from film CV measurements. Both reduction and oxidation of RingBDT(T3A)2 solution happened earlier than in film. Likely, the chromophore molecules assembled on the Pt surface in a way not conducive to electron transfer.
Mobility of pristine RingBDT(T3A)2 and BDT(T3A)2 films was measured by the space charge limited current (SCLC) and time of flight (TOF) methods. The sample preparation and measurement details are given in the experimental section. Chloroform and o-dichlorobenzene (o-DCB) were used for preparation of SCLC and TOF samples, respectively. The average SCLC hole mobilities of 1.52 × 10−3 and 6.225 × 10−4 cm2 V−1 s−1 were measured for films of RingBDT(T3A)2 (200 nm) and BDT(T3A)2 (80 nm), respectively. For TOF measurement, thicker films were able to be fabricated for RingBDT(T3A)2 (1500 nm) due to its better solution processability, while 450 nm films were obtained for BDT(T3A)2. The TOF charge carrier mobility of RingBDT(T3A)2 (8 × 10−3 to 8 × 10−4 cm2 V−1 s−1) was higher than that of BDT(T3A)2 (2 × 10−4 to 5 × 10−5 cm2 V−1 s−1) by 40–16 times (Fig. 9c). The mobilities of RingBDT(T3A)2 films measured by the two methods are close, while the TOF mobilities of BDT(T3A)2 films are 3 to 12 times smaller than the SCLC mobilities. The results indicates that carrier mobility in RingBDT(T3A)2 films is not only much higher, but also much less sensitive to choice of solvent used for film fabrication. The higher hole mobility of RingBDT(T3A)2 is attributed to the presence of less domain boundaries than in the crystalline BDT(T3A)2 films.
2.8 Solar cell devices
RingBDT(T3A)2 and BDT(T3A)2 chromophores were blended with PC60BM for fabrication of bulk heterojunction solar cells in the inverted device structure shown in Fig. 10a. The current–voltage characteristics under AM 1.5 are shown in Fig. 10b. The photovoltaic parameters of the best performing devices and the average power conversion efficiencies (PCE) of devices fabricated from the two blend materials under different processing conditions are listed in Table 2. The photovoltaic parameters of all solar cells are given in Tables S1–S5.† BDT(T3A)2:PC60BM cells of as-cast active layer showed a 2.54% PCE with a short circuit current density (Jsc) of 4.65 mA cm−2, an open circuit voltage (Voc) of 0.86 V and a fill factor (FF) of 0.63. RingBDT(T3A)2:PC60BM devices exhibited a slightly higher Jsc of 5.01 mA cm−2 but had a lower PCE (1.53%) due to a lower FF value (0.35). It is noted that chromophore:fullerene ratio 1:1 was found optimum for RingBDT(T3A)2, higher than that for BDT(T3A)2 (1:0.5), presumably due to its amorphous nature causing difficulty for phase separation of RingBDT(T3A)2 and PC60BM.
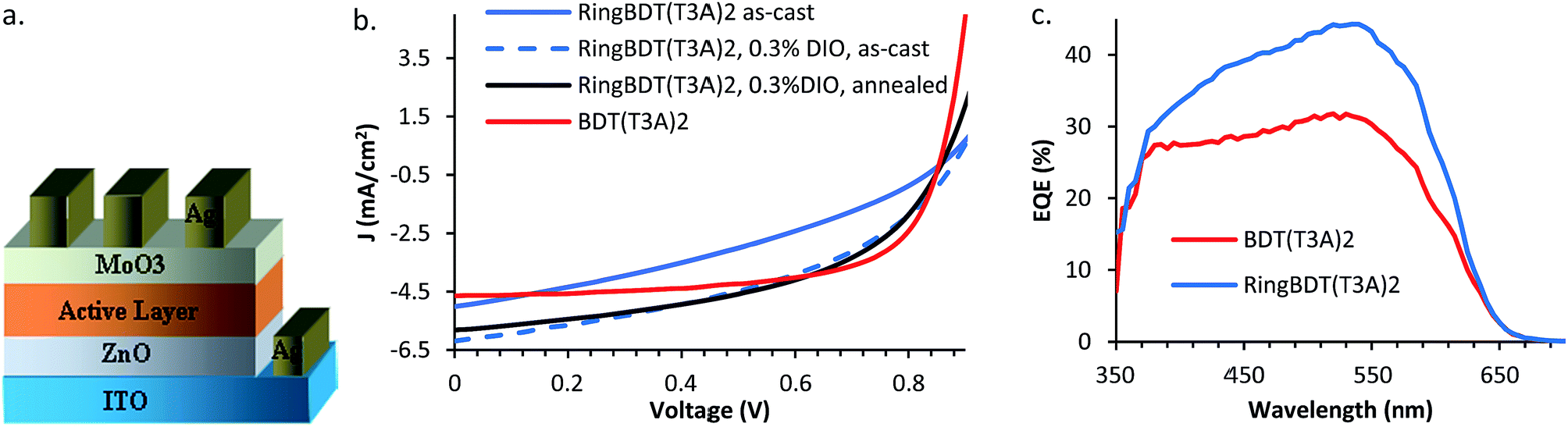 | ||
| Fig. 10 (a) Inverted solar cell device structure, (b) current density–voltage plot of RingBDT(T3A)2 and BDT(T3A)2 solar cells under AM 1.5, and (c) EQE of best performing devices. | ||
| Voc (V) | Jsc (mA cm−2) | Fill factor | Efficiency (%) max (average) | |
|---|---|---|---|---|
| BDT(T3A)2:PC60BM (as deposited) | 0.86 | 4.65 | 0.63 | 2.54 (2.23) |
| BDT(T3A)2:PC60BM (annealed) | 0.82 | 1.19 | 0.48 | 0.47 (0.38) |
| RingBDT(T3A)2:PC60BM (as deposited) | 0.87 | 5.01 | 0.35 | 1.53 (1.49) |
| RingBDT(T3A)2:PC60BMDIO (as deposited) | 0.88 | 6.19 | 0.43 | 2.34 (2.18) |
| RingBDT(T3A)2:PC60BMDIO (annealed) | 0.86 | 5.82 | 0.49 | 2.46 (2.24) |
Insufficient phase separation led to larger Jsc in RingBDT(T3A)2:PC60BM cells, but also negatively affected carrier life time and, in turn, reduced FF since it is known that FF depends on the mobility (μ)–lifetime (t) product (as well as thickness of the active layer).75 To improve FF of RingBDT(T3A)2:PC60BM devices, diiodooctane (DIO) was added at a level of 0.3 v% to chloroform. FF was improved by 23% to 0.43, Jsc was also improved by 23.6%, and PCE was improved to 2.34%. Further improvement was achieved by thermal annealing of the DIO devices at 100 °C for 10 minutes. A further gain of 14% was obtained for FF, accompanied by a minor (6%) loss in Jsc. The efficiency was raised to 2.46%, comparable to the best result (2.54%) of BDT(T3A)2:PC60BM devices. The overall enhancement in Jsc by DIO addition and annealing was further verified by the external quantum efficiency (EQE) spectra (Fig. 10c) of the best performing devices at short circuit conditions.
On the other hand, addition of DIO to BDT(T3A)2:PC60BM solution led to films that are severely rough to the eyes and devices with no PV performance. Thermal annealing at 100 °C for 10 minutes caused a dramatic decline in Jsc and a significant drop in FF, reducing the PCE from 2.54% to 0.46%. This result shows once again that thermal stability is a big problem for solar cell devices of highly crystalline small molecule materials.
The superior solution processability of RingBDT(T3A)2:PC60BM is also demonstrated in a series of devices of normal configuration: glass/ITO/PEDOT-PSS/donor:PC60BM/Ca/Al. With o-DCB as the solvent, which has a boiling point of 180 °C, much higher than that of chloroform (61 °C), similar efficiency (2.15%) was obtained, while decent films could not even be made from BDT(T3A)2:PC60BM. When chloroform was used, the best efficiency of BDT(T3A)2:PC60BM cells of the normal structure was only 0.63%. When 0.2 wt% of chlorobenzene was added to chloroform, the efficiency dropped to almost zero (0.015%). More detailed results of normal device structures are included in Tables S6 and S7.†
2.9 AFM and XRD study of chromophore:PC60BM blend films
To correlate the device performance with morphology, atomic force microscopy was performed on the active layers as shown in Fig. 11. The RingBDT(T3A)2:PC60BM topographic images show a smooth surface morphology with root mean square (RMS) roughness of 0.416 nm when casted from a chloroform solution. On the other hand, BDT(T3A)2:PCBM films showed a rough morphology with large domains of 20–30 nm in size and a RMS roughness of 6.98 nm.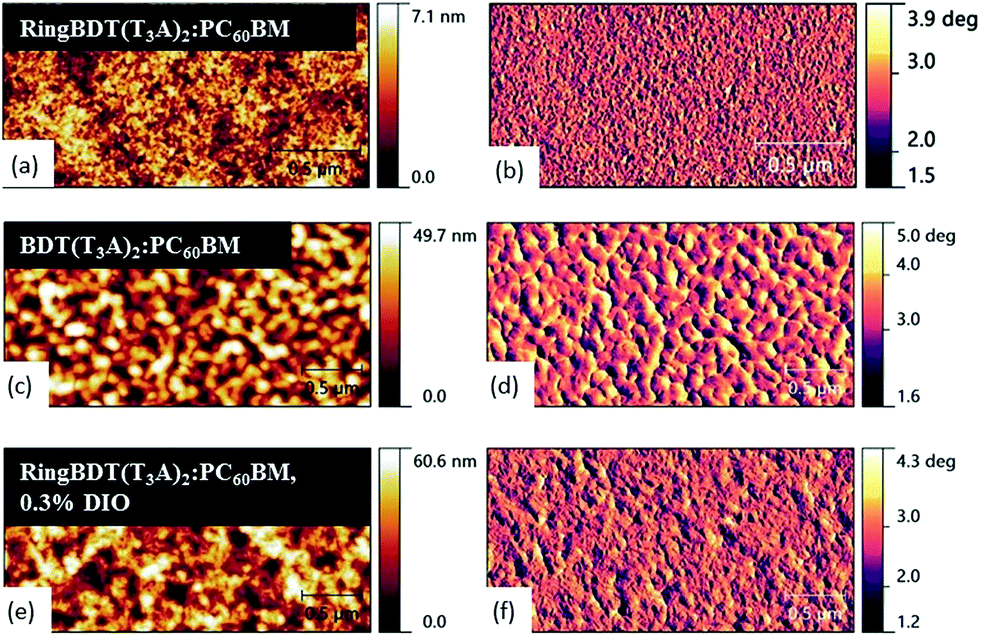 | ||
| Fig. 11 AFM topographic (left) and phase images (right) of RingBDT(T3A)2:PC60BM (without and with use of 0.3 v% DIO) and BDT(T3A)2:PC60BM films. Chloroform was used as the solvent. | ||
For both chromophores, the surface morphology did not change much with fullerene addition as compared with the surface morphology of pristine films in Fig. 6. With the addition of 0.3 v% DIO to the solution of RingBDT(T3A)2:PC60BM, slightly larger domains are seen in both topography and phase images and the RMS roughness of films increased to 6.01 nm. Aggregation of RingBDT(T3A)2 is also confirmed by the change in UV-vis absorption spectral profile upon addition of DIO (Fig. S24†).
XRD measurements of RingBDT(T3A)2:PCBM films casted from the chloroform–DIO solution did not show any peaks before and after annealing at 100 °C for 10 minutes (figures not presented), in consistence with the effect of annealing on RingBDT(T3A)2 thin films (Fig. 5b).
2.10 Photo-CELIV charge carrier mobility and density
Although the RingBDT(T3A)2 pristine films had a much higher hole mobility than BDT(T3A)2, its optimum PV efficiency was only similar to that of BDT(T3A)2. To understand the reason behind the unexpected result, charge carrier mobility and density in the solar cells were investigated using the photo-generated charge extraction by linearly increasing voltage (Photo-CELIV) technique.76,77 Fig. 12a shows the Photo-CELIV curves of the best RingBDT(T3A)2 and BDT(T3A)2 solar cells and the extracted charge carrier density (ηext) with varying delay time. The mobility and charge carrier densities were calculated as explained in our previous papers,77,78 and are tabulated in Table 3.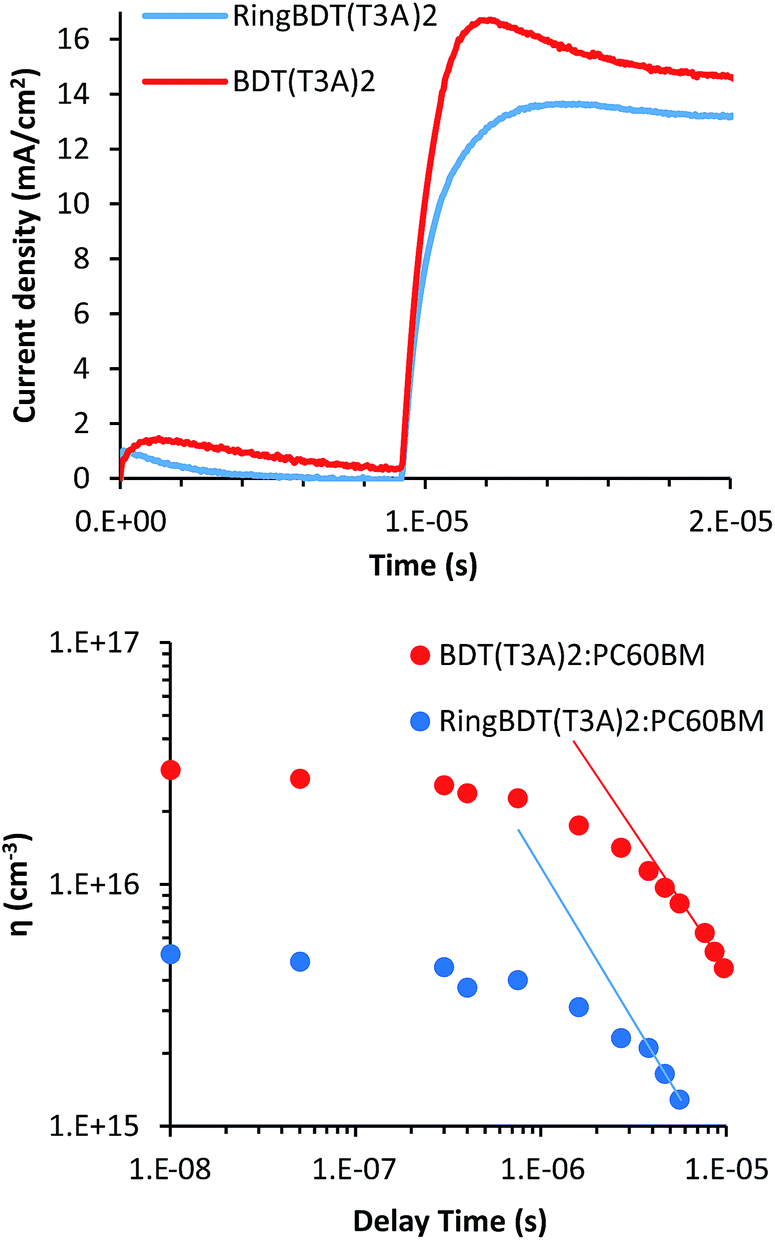 | ||
| Fig. 12 The Photo-CELIV curves of best RingBDT(T3A)2 and BDT(T3A)2 solar cells and the extracted charge carrier density (ηext) with varying delay times. | ||
BDT(T3A)2:PC60BM devices have higher current density and shorter tmax (the time at which the current reaches maximum) compared to RingBDT(T3A)2:PC60BM. The shorter tmax means that the lower bias/field was needed to extract the maximum amount of charge carriers, a result of higher mobility. The charge carrier mobilities of 1.44 × 10−5 and 1.76 × 10−5 cm2 V−1 s−1 were calculated for RingBDT(T3A)2:PC60BM and BDT(T3A)2:PC60BM devices at 8 μs delay time, respectively. Further, the integrated extracted charge carrier densities (ηext) from the Photo-CELIV data at different delay times are plotted in Fig. 12b. BDT(T3A)2:PC60BM devices show an order of magnitude higher ηexts than those of RingBDT(T3A)2:PCBM devices, and both devices show a power law decay, indicative of bimolecular recombination in the devices. The slopes of linear regions at higher delay times are −1.01 and −1.21 cm−3 s−1 for BDT(T3A)2 and RingBDT(T3A)2 devices, respectively. The higher slope of RingBDT(T3A)2 device indicates higher recombination rate of photo-generated carriers.
Albeit the higher mobility in pristine films, RingBDT(T3A)2 exhibits slightly lower mobility when blended with PC60BM. Better mixing of RingBDT(T3A)2 and PC60BM leads to higher interfacial area for exciton dissociation and free charge carrier generation, but also leads to higher bimolecular recombination as the probability of free electron and hole meeting each other increases. The higher recombination rate explains the observed lower mobility and fill factor in devices.
3. Conclusions
A novel ring-protected small molecule chromophore, RingBDT(T3A)2, has been developed to address the low morphological stability issue of small molecule solar cells. In single crystals, its precursor 8 formed π-dimers. 1H NMR and UV-vis absorption studies of solutions showed that RingBDT(T3A)2 forms dimers in chloroform. DSC study showed that upon cooling RingBDT(T3A)2 melt crystallizes much slower than that of the ringless chromophore, BDT(T3A)2, but RingBDT(T3A)2 solid crystallize rather fast and complete upon heating at around 110 °C. XRD analysis showed that RingBDT(T3A)2 was able to crystallize in thick films (2.5–3.5 μm) upon annealing at 100 °C or 125 °C, but show no or little sign of crystallinity in thin films (200 nm) after annealing at the same temperatures. Addition of solvent additive and thermal annealing both enhanced π–π stacking but did not result in formation of crystalline structure. Despite the amorphous nature of its films, SCLC hole mobility of RingBDT(T3A)2 was 144% higher than that of BDT(T3A)2 in thin films (200–80 nm). BDT(T3A)2 crystallized fast when casted into thin films and possessed large rough domains, while RingBDT(T3A)2 gave smooth surface. When blended with PC60BM, RingBDT(T3A)2 exhibited higher photocurrent due to better mixing with the acceptor. However, it also caused faster charge recombination, resulting in a lower fill factor. FF of RingBDT(T3A)2 cells was improved significantly from 0.35 to 0.49 by using a solvent additive (DIO) and thermal annealing, while the performance of BDT(T3A)2 solar cells dropped dramatically with thermal annealing under the same condition (100 °C 10 min). Charge carrier mobility and density in both blend films were studied using the Photo-CELIV technique. RingBDT(T3A)2:PC60BM blend had lower mobility and lower extracted charge carrier density due to higher bimolecular recombination. Overall, ring protection is a promising strategy for development of small molecules with good morphological stability for solar cells. Future research is needed to improve phase separation of ring-protected chromophores and electron acceptors.4. Experimental section
PC60BM was obtained from Nano-C. Zinc oxide sol–gel was prepared according procedures reported.79 All other chemicals were obtained from either Sigma Aldrich or Fisher Scientific and used without further purification.4.1 2,5-Bis(2-oxo-ethylsulfanyl)-1,4-(1,12-dodecylenedioxy)benzene (4)
A 2 L 3-neck flask, equipped with a thermometer, was flushed with N2 gas, charged with 1 (59.51 g, 137.05 mmol) and diethyl ether (595 mL), and cooled in a −60 °C bath. BuLi (2.31 mol eq., 125.2 mL) was added when the internal temperature was lowered to −35 °C. Addition rate was kept low so that the temperature dropped to −65 °C at the end of addition. The reaction was monitored by NMR to assure that the dilithiation reaction was complete. 1,2-Bis(2,2-diethoxyethyl)disulfide (2)80 (2.93 eq., 120 g, 96.5%, homemade) was added to the reaction solution at −65 °C over 1 h. The reaction was allowed to warm up to room temperature. Ether was removed by rotary evaporation under 500 mbar. Hexanes (500 mL) and water (100 mL) were added to dissolve the residue. The aqueous phase was separated and extracted with hexanes (50 mL). Hexane extracts were combined and frozen for 1 h to remove hexane-insoluble impurities. The clear solution was decanted into a 2 L flask and condensed by rotary evaporation at 250 mbar to afford a yellow oil (141.5 g). A mixture of the crude intermediate product (3), acetone (2100 g), water (360 g) and 2 N HCl (360 g) was stirred at 57 °C for 40 min to hydrolyze the acetal groups. The mixture was then neutralized with NaHCO3 powder. After acetone was removed by rotary evaporation, the crude oil was taken up by hexanes. The hexane solution was dried with MgSO4 and condensed by rotary evaporation under 250 mbar. The residue was vacuumed with an oil pump at 90 °C to remove volatile impurities. The product was used in the next reaction without purification. 1H NMR (CDCl3): δ 0.94 (m, 8H), 1.04–1.24 (m, 4H), 1.24–1.48 (m, 4H), 1.5–1.67 (m, 2H), 1.68–1.84 (m, 2H), 3.49 (dd, J = 15.4 & 2.9 Hz, 2H), 3.65 (dd, J = 15.4 & 3.5 Hz, 2H), 6.86 (s, 2H), 9.54 (t, 2H). The dialdehyde product is not stable, and was not submitted for HRMS analysis.4.2 4,8-(1,12-Dodecylenedioxy)benzo[1,2-b:4,5-b′]dithiophene (5)
This reaction was performed in small scales. A typical run: a mixture of crude 4 (14 g), chlorobenzene (1.5 L) and Amberlyst 15 (300 mL) was boiled under reflux with N2 protection for 2 h. The solvent was removed by rotary evaporation. Crude products of all runs were combined and purified using silica gel column chromatography with EtOAc/hexanes (1/64 vol) as the eluent. The pure fractions were combined and condensed. The product crystallized from the residue solutions after rotary evaporation. Yield 8.52 g, 16% for three steps from 1 and 2. 1H NMR (CDCl3): δ 0.71 (m, 8H), 0.97 (m, 4H), 1.27 (m, 4H), 1.64 (m, 4H), 4.42 (m, 2H), 4.64 (m, 2H), 7.35 (d, J = 5.6 Hz), 7.51 (d, J = 5.6 Hz). 13C NMR (CDCl3): δ 24.22, 27.16, 27.24, 27.81, 28.75, 71.96, 120.60, 125.73, 129.46, 131.24, 143.60 ppm. HRMS calculated for C22H28O2S2: 388.153. Found: 388.153.4.3 2,6-Bis(trimethylstannyl)-4,8-(1,12-dodecylenedioxy)benzo[1,2-b:4,5-b′]dithiophene (6)
In a N2 glovebox, 2.5 M BuLi (2.1 eq., 10.6 mL) was added in one shot to a solution of 5 (4.893 g, 12.59 mmol) in THF (150 mL) at RT. The solution turned cloudy immediately. Two minutes later, trimethylstannyl chloride (26.69 mmol, 5.319 g) was added at once. The solution became clear in 10 s. The reaction flask was taken out of glovebox. Saturated aqueous NaHCO3 (3 mL) was added to the reaction mixture. The solvent was removed by rotary evaporation at 100 mbar. The residue was dissolved by hexanes. The hexane solution was dried with anhydrous Na2CO3, condensed, and vacuum dried to afford 9.03 g, yield: 100%. 1H NMR spectrum (in the ESI†) shows that there is only trace amount of impurity in the aromatic region. Purification was attempted using silica gel chromatography without success because the Rf values of 5, 6 and the mono-stannylated product are nearly same even in hexanes. 1H NMR (CDCl3): δ 0.44 (s, 18H), 0.66 (m, 4H), 0.74 (m, 4H), 0.97 (m, 4H), 1.28 (m, 4H), 1.64 (m, 4H), 4.42 (m, 2H), 4.70 (m, 2H), 7.56 (s, 2H). 13C NMR (CDCl3): δ −7.32, 25.46, 28.34 (two signals overlapped), 28.79, 29.95, 72.67, 129.30, 133.74, 134.25, 140.97, 143.22 ppm. HRMS calculated for C28H44O2S2Sn2: 716.083. Found: 716.080.4.4 Compound 8
A mixture of 6 (2.816 mmol, 2.012 g), 781 (5.628 mmol, 2.000 g), Pd(PPh3)4 (0.056 mmol, 64.0 mg) and THF (∼3 mL) in a pressure reactor was stirred at 120 °C for 30 hours. The crude product was purified with a silica gel column using chloroform as eluent. Crystallization from EtOAc (solubility < 1 mg/100 mL) afforded 2.13 g product with a yield of 80.7%. 1H NMR (CDCl3): δ 0.89–0.71 (m, 8H), 1.03 (m, 4H), 1.33 (m, 4H), 1.71 (m, 4H), 4.44 (m, 2H), 4.66 (m, 2H), 7.16 (d, 2H, J = 3.9 Hz), 7.17 (d, 2H, J = 3.8 Hz), 7.23 (d, 2H, J = 3.8 Hz), 7.24 (d, 2H, J = 4.1 Hz), 7.29 (d, 2H, J = 3.8 Hz), 7.54 (s, 2H), 7.67 (d, 2H, J = 3.9 Hz), 9.86 (s, 2H). 13C NMR (CDCl3): δ 182.42, 146.61, 143.12, 141.71, 138.56, 137.40, 137.13, 136.35, 135.80, 134.87, 132.24, 128.67, 127.03, 125.07, 125.17, 124.86, 124.16, 116.66, 72.09, 28.83, 27.81, 27.32, 27.21, 24.31. Anal. calcd for C48H40O4S8: C, 61.50; H, 4.30. Found: C, 61.25; H, 4.23.4.5 RingBDT(T3A)2
A mixture of 8 (0.47 mmol, 440 mg), 982 (1.17 mmol, 162.6 mg), chloroform (8.0 mL) and triethylamine (39 mg) in a 32 mL vial was heated with stirring in a 60 °C oil bath for 16 h. The crude product was purified with a silica gel column using chloroform as eluent. Crystallization from EtOAc afforded 460 mg product with a yield of 79%. 1H NMR (CDCl3): δ 0.62–0.83 (m, 8H), 0.95–1.05 (m, 10H including a triplet at 0.91 of 6H – two CH3), 1.2–1.5 (m, 16H including 12H from underlined CH2 groups in two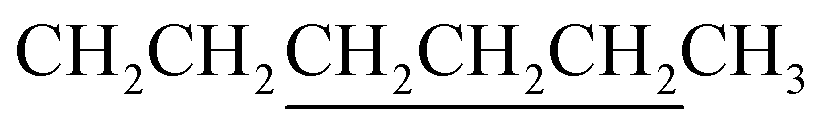 ), 1.5–2.0 (m, 8H including 4H from underlined CH2 groups in two
), 1.5–2.0 (m, 8H including 4H from underlined CH2 groups in two 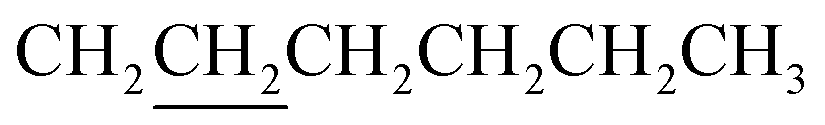 ), 4.23 (m, 4H, –OCH2), 4.39 (m, 2H), 4.61 (m, 2H), 7.05 (d, J = 3.8 Hz), 7.13 (d, 2H, 3.8 Hz), 7.15 (d, 2H, 4.1 Hz), 7.23 (d, 2H, J = 3.8 Hz), 7.34 (s, 2H), 7.59 (d, 2H, J = 4.0 Hz), 8.16 (s, 2H). 13C NMR (CDCl3): δ 162.97, 148.92, 145.80, 142.99, 139.05, 138.93, 137.24, 136.18, 135.69, 134.41, 134.28, 132.20, 128.63, 127.29, 125.95, 125.10, 124.83, 124.21, 116.61, 116.04, 97.55, 72.04, 66.60, 31.44, 28.85, 28.55, 27.80, 27.32, 27.22, 25.52, 24.33, 22.56, 14.03. Anal. calcd for C66H66N2O6S8: C, 63.94; H, 5.37. Found: C, 63.67; H, 5.38.
), 4.23 (m, 4H, –OCH2), 4.39 (m, 2H), 4.61 (m, 2H), 7.05 (d, J = 3.8 Hz), 7.13 (d, 2H, 3.8 Hz), 7.15 (d, 2H, 4.1 Hz), 7.23 (d, 2H, J = 3.8 Hz), 7.34 (s, 2H), 7.59 (d, 2H, J = 4.0 Hz), 8.16 (s, 2H). 13C NMR (CDCl3): δ 162.97, 148.92, 145.80, 142.99, 139.05, 138.93, 137.24, 136.18, 135.69, 134.41, 134.28, 132.20, 128.63, 127.29, 125.95, 125.10, 124.83, 124.21, 116.61, 116.04, 97.55, 72.04, 66.60, 31.44, 28.85, 28.55, 27.80, 27.32, 27.22, 25.52, 24.33, 22.56, 14.03. Anal. calcd for C66H66N2O6S8: C, 63.94; H, 5.37. Found: C, 63.67; H, 5.38.
4.6 BDT(T3A)2
A mixture of 1010 (0.201 mmol, 0.291 g), 982 (0.615 mmol, 85.4 mg), chloroform (8.0 mL) and triethylamine (39 mg) in a 32 mL vial was heated with stirring in a 60 °C oil bath for 19 h. EtOAc (3 mL) was added to the reaction mixture. The solution was boiled for 1 minute, and then was allowed to cool down to RT. Product was collected by filtration and washed with EtOAc. Yield: 317 mg, 90.1%. 1H NMR (CDCl3): δ 0.88 (m, 18H), 0.99 (t, 6H), 1.07 (t, 6H), 1.20–1.50 (m, 60H from CH and non-terminal CH2 groups in the alkyl groups), 1.73 (m, 20H from β-CH2 units in alkyl groups on thiophenes and acceptors, and γ-CH2 units in alkyl groups on the BDT), 1.85 (m, 2H from the two CH units), 2.75–2.95 (m, 8H from the α-CH2 on thiophenes), 4.19 (d, J = 5.2 Hz, 4H from the α-CH2 on the BDT), 4.29 (t, J = 6.7 Hz, 4H, –OCH2), 7.14 (s, 2H), 7.16 (d, 2H, J = 3.9 Hz), 7.31 (d, 2H, 3.9 Hz) (from the two middle thiophenes), 7.46 (s, 2H), 7.59 (s, 2H), 8.19 (s, 2H). 13C NMR (CDCl3): 163.11, 145.91, 144.10, 141.59, 141.25, 140.75, 140.54, 138.31, 136.08, 135.75, 134.35, 132.97, 132.47, 130.19, 129.20, 128.35, 128.23, 126.34, 116.22, 116.01, 97.72, 66.56, 40.70, 31.91, 31.87, 31.40, 30.48, 30.44, 30.23, 29.72, 29.68, 29.66, 29.56, 29.48, 29.42, 29.41, 29.31, 29.27, 29.25, 28.52, 25.48, 23.88, 23.24, 22.69, 22.68, 22.53, 14.29, 14.13, 14.12, 14.01, 11.40. Anal. calcd for C102H140N2O6S8: C, 70.14; H, 8.08. Found: C, 69.97; H, 7.97.4.7 Cyclic voltammetry
Cyclic voltammetry measurements were performed on an eDAQ Potentiostat System (ER466 plus software EChem 2.1.5) equipped with platinum auxiliary and working electrodes and a silver reference electrode in a CH3CN solution of AgNO3 (0.01 M) and tetrabutylammonium hexafluorophosphonate, TBA-HFP (0.1 M). Samples were dissolved in chloroform and then dip coated onto the Pt wire working electrode. The electrodes are housed in an 8 mL vial half-filled with TBA-HFP/acetonitrile solution (0.1 M). The solution was N2-purged and a scan rate of 100 mV s−1 was used. Between measurements, the surface of the electrodes were cleaned. Ferrocene (2 mM in the electrolyte solution) was used as an internal reference standard.4.8 Optical and XRD
X-ray diffraction (XRD) and UV-Visible optical absorption spectra were taken on thin films casted on ITO substrates coated with ZnO. Agilent 8853 spectrophotometer was used to record optical absorption spectra. ITO/ZnO films were taken as blank for optical measurements. Rigaku SmartLab diffractometer (Cu Kα radiation, λ = 1.5406 Å) was used to record XRD spectra.4.9 Morphology
Atomic force microscopy topography and phase images were taken in tapping mode operation with Agilent 5500 SPM using a silicon cantilever having resonant frequency ∼290 kHz and force constant ∼40 N m−1 (Budget Sensors TAP 300Al). The imaging was done under net attractive force mode. All images were analyzed using Gwyddion software.4.10 Time of flight (TOF) mobility
TOF devices were fabricated with a structure of indium tin oxide (ITO)/RingBDT(T3A)2 (1500 nm), BDT(T3A)2 (450 nm)/N,N′-bis(naphthalen-1-yl)-N,N′-bis(phenyl)-benzidine (NPB) (100 nm)/Al (40 nm). RingBDT(T3A)2 and BDT(T3A)2 were dissolved in o-dichlorobenzene (15 g L−1) and the solutions were spin-coated on ITO-glass substrates at 600 rpm for 60 seconds and then dried for 10 minutes. The same process was repeated a few times to obtain the desired film thickness. Then, NPB and Al electrode were deposited by thermal evaporation at a rate of 2 Å s−1, where NPB layer was used as a charge generation layer. The specimen was encapsulated using an epoxy resin and a clean glass to avoid oxygen and moisture. In TOF measurement, an optical pulse incident on the material creates photo-generated carriers to drift across the sample to the charge-collecting electrode under an applied electric field.83 A digital oscilloscope (Tektronix TDS3032B) was used to capture the voltage across a current sensing resistor to obtain the transient curve. The typical transient curves of TOF under applied biases were plotted in a double logarithmic scale to extract the transit time. The hole mobility of the chromophore can be obtained from the transit time, which is determined by the onset of the tail.4.11 Space charge limited current (SCLC) mobility measurement
Devices for SCLC were fabricated with hole only interface. The device structure used was ITO/PEDOT/chromophore/MoO3/Ag. PEDOT:PSS was spun at 5000 rpm for 30 seconds and annealed at 140 °C for 10 minutes. The chromophores were coated at 2000 rpm for 30 seconds. The hole mobilities (μh) were calculated using Mott–Gurney law as followswhere ε is dielectric permittivity, V is the applied voltage, d is the thickness and J is the measured current density.
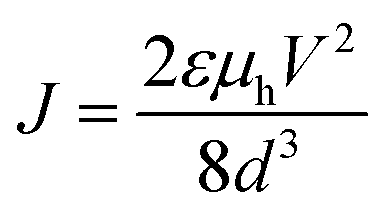
4.12 Cell fabrication
ITO coated glass substrates (sheet resistance ∼ 10 Ω □−1) were sequentially cleaned with detergent, DI water, acetone and IPA by ultrasonication for 20 minutes each. The substrates were dried with nitrogen gas and then were subjected to oxygen plasma treatment for 25 minutes to make the surface hydrophilic. ZnO sol–gel was then spin-coated on plasma cleaned ITO at 4500 rpm for 60 seconds and annealed at 200 °C for 30 minutes. The substrates were then transferred to a nitrogen glove box where the active layer was coated at 2000 rpm for 30 seconds. Blend solutions were made by mixing appropriate weight ratios of small molecule and fullerene in chloroform. The concentration of donor was kept 8 mg mL−1 for all chromophores. Certain films were annealed in glove box at 100 °C for 10 minutes. The active layer films were then transferred to thermal evaporator integrated within the glove box. 10 nm of MoO3 and 80 nm of Ag were deposited under a vacuum of 10−6 mbar.4.13 Device characterization
Current density–voltage spectroscopy was performed using Agilent 4155C semiconductor parameter analyzer. A xenon arc lamp with an AM 1.5 filter was used as the light source. The light intensity was calibrated using an NREL calibrated photodetector. External quantum efficiency spectra was taken using the same lamp integrated with a Newport monochromator controlled using a Labview program. Photo-charge extraction by linearly increasing voltage (Photo-CELIV) measurements were done using a custom written Labview program. A nanosecond laser source (OBB 401 Dye laser) was incident on the solar cell which was kept at open circuit condition. A voltage ramp of 2 V (80000 kHz) was provided to the cells connected in reverse bias with user given delay time at a ramp frequency of 80 kHz. Photo-CELIV spectra was recorded on an Agilent MSO7034B oscilloscope.
Acknowledgements
This material is based upon work supported by the National Science Foundation/EPSCoR (Award No. 0903804), the NASA EPSCoR (Award No. NNX13AD31A), and South Dakota Board of Regents Competitive Research Grant Award 2013-10-06.References
- I. Etxebarria, J. Ajuria and R. Pacios, Org. Electron., 2015, 19, 34–60 CrossRef CAS.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- X. Ouyang, R. Peng, L. Ai, X. Zhang and Z. Ge, Nat. Photonics, 2015, 9, 520–524 CrossRef CAS.
- Y. Liu, Z. A. Page, T. P. Russell and T. Emrick, Angew. Chem., Int. Ed., 2015, 54, 11485–11489 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2016, 24, 905–913 Search PubMed.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- J. Zhou, X. Wan, Y. Liu, Y. Zuo, Z. Li, G. He, G. Long, W. Ni, C. Li, X. Su and Y. Chen, J. Am. Chem. Soc., 2012, 134, 16345–16351 CrossRef CAS PubMed.
- J. Zhou, Y. Zuo, X. Wan, G. Long, Q. Zhang, W. Ni, Y. Liu, Z. Li, G. He, C. Li, B. Kan, M. Li and Y. Chen, J. Am. Chem. Soc., 2013, 135, 8484–8487 CrossRef CAS PubMed.
- S. Mukherjee, C. M. Proctor, J. R. Tumbleston, G. C. Bazan, T.-Q. Nguyen and H. Ade, Adv. Mater., 2015, 27, 1105–1111 CrossRef CAS PubMed.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS PubMed.
- W. Ma, J. R. Tumbleston, C. W. L. Ye, J. Hou and H. Ade, Adv. Mater., 2014, 26, 4234–4241 CrossRef CAS PubMed.
- J. Yuan, H. Dong, M. Li, X. Huang, J. Zhong, Y. Li and W. Ma, Adv. Mater., 2014, 26, 3624–3630 CrossRef CAS PubMed.
- I. T. Lima, C. Risko, S. G. Aziz, D. A. d. S. Filho and J.-L. Bredas, J. Mater. Chem. C, 2014, 2, 8873–8879 RSC.
- K. R. Graham, C. Cabanetos, J. P. Jahnke, M. N. Idso, A. E. Labban, G. O. N. Ndjawa, T. Heumueller, K. Vandewal, A. Salleo, B. F. Chmelka, A. Amassian, P. M. Beaujuge and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 9608–9618 CrossRef CAS PubMed.
- P. Liu, K. Zhang, F. Liu, Y. Jin, S. Liu, T. P. Russell, H.-L. Yip, F. Huang and Y. Cao, Chem. Mater., 2014, 26, 3009–3017 CrossRef.
- J. R. Tumbleston, B. A. Collins, L. Yang, A. C. Stuart, E. Gann, W. Ma, W. You and H. Ade, Nat. Photonics, 2014, 8, 385–391 CrossRef CAS.
- T. S. v. d. Poll, J. A. Love, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646–3649 CrossRef PubMed.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- A. B. Tamayo, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 11545–11551 CAS.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed.
- J. A. Love, C. M. Proctor, J. Liu, C. J. Takacs, A. Sharenko, T. S. v. d. Poll, A. J. Heeger, G. C. Bazan and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 27, 5019–5026 CrossRef.
- W. Ni, M. Li, X. Wan, H. Feng, B. Kan, Y. Zuo and Y. Chen, RSC Adv., 2014, 4, 31977–31980 RSC.
- Z. Li, G. He, X. Wan, Y. Liu, J. Zhou, G. Long, Y. Zuo, M. Zhang and Y. Chen, Adv. Energy Mater., 2012, 2, 74–77 CrossRef CAS.
- X. Wan, Y. Liu, F. Wang, J. Zhou, G. Long and Y. Chen, Org. Electron., 2013, 14, 1562–1569 CrossRef CAS.
- Y. Liu, Y. M. Yang, C.-C. Chen, Q. Chen, L. Dou, Z. Hong, G. Li and Y. Yang, Adv. Mater., 2013, 25, 4657–4662 CrossRef CAS PubMed.
- Y. Chen, Z. Du, W. Chen, L. Han, Q. Liu, M. Sun and R. Yang, Synth. Met., 2014, 187, 24–29 CrossRef CAS.
- Z. Yi, W. Ni, Q. Zhang, M. Li, B. Kan, X. Wan and Y. Chen, J. Mater. Chem. C, 2014, 2, 7247–7255 RSC.
- K. Sun, Z. Xiao, E. Hanssen, M. F. G. Klein, H. H. Dam, M. Pfaff, D. Gerthsen, W. W. H. Wong and D. J. Jones, J. Mater. Chem. A, 2014, 2, 9048–9054 CAS.
- J. Zhou, X. Wan, Y. Liu, G. Long, F. Wang, Z. Li, Y. Zuo, C. Li and Y. Chen, Chem. Mater., 2011, 23, 4666–4668 CrossRef CAS.
- G. Li, V. Shrotriya, Y. Yao and Y. Yang, J. Appl. Phys., 2005, 98, 043704 CrossRef.
- M. Shin, H. Kim, J. Park, S. Nam, K. Heo, M. Ree, C.-S. Ha and Y. Kim, Adv. Funct. Mater., 2010, 20, 748–754 CrossRef CAS.
- Y. Shirot, J. Mater. Chem., 2005, 15, 75–93 RSC.
- K. Naito and A. Miura, J. Phys. Chem. C, 1993, 97, 6240–6248 CrossRef CAS.
- Y. Ie, M. Endou, S. K. Lee, R. Yamada, H. Tada and Y. Aso, Angew. Chem., Int. Ed., 2011, 50, 11980–11984 CrossRef CAS PubMed.
- Y. Ie, M. Endou, A. Han, R. Yamada, H. Tada and Y. Aso, Pure Appl. Chem., 2012, 84, 931–943 CrossRef CAS.
- C. Zhao, T. Sakurai, S. Yoneda, S. Seki, M. Sugimoto, C. Oki, M. Takeuchi and K. Sugiyasu, Chem.–Asian J., 2016, 2284–2290 CrossRef CAS PubMed.
- C. Pan, K. Sugiyasu, J. Aimi, A. Sato and M. Takeuchi, Angew. Chem., Int. Ed., 2014, 53, 8870–8875 CrossRef CAS PubMed.
- P. D. Frischmann and F. Würthner, Org. Lett., 2013, 15, 4674–4677 CrossRef CAS PubMed.
- J. Lim and T. M. Swager, Angew. Chem., Int. Ed., 2010, 49, 7486–7488 CrossRef CAS PubMed.
- M. Morales-Vidal, P. G. Boj, J. M. Villalvilla, J. A. Quintana, Q. Yan, N.-T. Lin, X. Zhu, N. Ruangsupapichat, J. Casado, H. Tsuji, E. Nakamura and M. A. Díaz-García, Nat. Commun., 2015, 6, 8458 CrossRef CAS PubMed.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
- K. Sugiyasu and M. Takeuchi, Chem.–Eur. J., 2009, 15, 6350–6362 CrossRef CAS PubMed.
- N. Stingelin-Stutzmann, E. Smits, H. Wondergem, C. Tanase, P. Blom, P. Smith and D. d. Leeuw, Nat. Mater., 2005, 4, 601–606 CrossRef CAS PubMed.
- C. Lindqvist, J. Bergqvist, O. Bäcke, S. Gustafsson, E. Wang, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Appl. Phys. Lett., 2014, 104, 153301 CrossRef.
- Y. Santo, I. Jeon, K. S. Yeo, T. Nakagawa and Y. Matsuo, Appl. Phys. Lett., 2013, 103, 073306 CrossRef.
- N.-X. Hu, S. Xie, Z. Popovic, B. Ong, A.-M. Hor and S. Wang, J. Am. Chem. Soc., 1999, 121, 5097–5098 CrossRef CAS.
- J. B. Sherman, C.-Y. Chiu, R. Fagenson, G. Wu, C. J. Hawker and M. L. Chabinyc, MRS Commun., 2015, 5, 447–452 CrossRef CAS.
- C. Zhang, L. Zhang, S. J. Benight, B. C. Olbricht, L. E. Johnson, B. H. Robinson, R. A. Norwood and L. R. Dalton, Proc. SPIE, 2013, 8827, 4 CrossRef.
- P. S. Kalsi, in Stereochemistry Conformation and Mechanism, New Age International, 6th edn, 2006 Search PubMed.
- M. H. Yoon, A. Facchetti, C. E. Stern and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 5792–5801 CrossRef CAS PubMed.
- X. Chi, D. Li, H. Zhang, Y. Chen, V. Garcia, C. Garcia and T. Siegrist, Org. Electron., 2008, 9, 234–240 CrossRef CAS.
- M. M. Safont-Sempere, P. Osswald, K. Radacki and F. Würthner, Chem.–Eur. J., 2010, 16, 7380–7384 CrossRef CAS PubMed.
- M. M. Safont-Sempere, P. Osswald, M. Stolte, M. Grüne, M. Renz, M. Kaupp, H. B. K. Radacki and F. Würthner, J. Am. Chem. Soc., 2011, 133, 9580–9591 CrossRef CAS PubMed.
- J. Peet, C. Soci, R. C. Coffin, T. Q. Nguyen, A. Mikhailovsky, D. Moses and G. C. Bazana, Appl. Phys. Lett., 2006, 89, 252105 CrossRef.
- P. Cheng, Q. Shi, Y. Lin, Y. Li and X. Zhan, Org. Electron., 2013, 14, 599–606 CrossRef CAS.
- L. Yuan, Y. Zhao, K. Lu, D. Deng, W. Yan and Z. Wei, J. Mater. Chem. C, 2014, 2, 5842–5849 RSC.
- Q. Liu, Z. Du, W. Chen, L. Sun, Y. Chen, M. Sun and R. Yang, Synth. Met., 2013, 178, 38–43 CrossRef CAS.
- H. Bai, P. Cheng, Y. Wan, L. Ma, Y. Li, D. Zhu and X. Zhan, J. Mater. Chem. A, 2014, 2, 778–784 CAS.
- C. Zhang, T. Matos, R. Li, S.-S. Sun, J. E. Lewis, J. Zhang and X. Jiang, Polym. Chem., 2010, 1, 663–669 RSC.
- M. Shahid, R. S. Ashraf, E. Klemm and S. Sensfuss, Macromolecules, 2006, 39, 7844–7853 CrossRef CAS.
- Y. Xia, J. Luo, X. Deng, X. Li, D. Li, X. Zhu, W. Yang and Y. Cao, Macromol. Chem. Phys., 2006, 207, 511–520 CrossRef CAS.
- C. Zhang, T. H. Nguyen, J. Sun, R. Li, S. Black, C. E. Bonner and S.-S. Sun, Macromolecules, 2009, 42, 663–670 CrossRef CAS.
- Z. Lu, C. Li, T. Fang, G. Li and Z. Bo, J. Mater. Chem. A, 2013, 1, 7657–7665 CAS.
- V. Tamilavana, M. Song, J.-W. Kang and M. H. Hyun, Synth. Met., 2013, 176, 96–103 CrossRef.
- Y. Chen, Y. Yan, Z. Du, X. Bao, Q. Liu, V. A. L. Roy, M. Sun, R. Yang and C. S. Lee, J. Mater. Chem. C, 2014, 2, 3921–3927 RSC.
- Y. J. Kim, J. Y. Baek, J.-j. Ha, D. S. Chung, S.-K. Kwon, C. E. Park and Y.-H. Kim, J. Mater. Chem. C, 2014, 2, 4937–4946 RSC.
- Y. Kim, C. E. Song, A. Cho, J. Kim, Y. Eom, J. Ahn, S.-J. Moon and E. Lim, Mater. Chem. Phys., 2014, 143, 825–829 CrossRef CAS.
- R. L. Schwiderski and S. C. Rasmussen, Synth. Met., 2014, 193, 58–63 CrossRef CAS.
- X.-J. Wang, L. Wang, J.-J. Wang and T. Chen, J. Appl. Polym. Sci., 2006, 101, 515–523 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- D. Gupta, S. Mukhopadhyay and K. S. Narayan, Sol. Energy Mater. Sol. Cells, 2010, 94, 1309–1313 CrossRef CAS.
- K. Genevicius, R. Osterbacka, G. Juska, K. Arlauskas and H. Stubb, Synth. Met., 2003, 137, 1407–1408 CrossRef CAS.
- S. Venkatesan, E. C. Ngo, Q. Chen, A. Dubey, L. Mohammad, N. Adhikari, A. F. Mitul and Q. Qiao, Nanoscale, 2014, 6, 7093–7100 RSC.
- S. Venkatesan, J. Chen, E. C. Ngo, A. Dubey, D. Khatiwada, C. Zhang and Q. Qiao, Nano Energy, 2015, 12, 457–467 CrossRef CAS.
- S. Venkatesan, N. Adhikari, J. Chen, E. C. Ngo, A. Dubey, D. W. Galipeau and Q. Qiao, Nanoscale, 2014, 6, 1011–1019 RSC.
- J. T. Henssler and A. J. Matzger, Org. Lett., 2009, 11, 3144–3147 CrossRef CAS PubMed.
- Y. Wei, B. Wang, W. Wang and J. Tian, Tetrahedron Lett., 1995, 36, 665–668 CrossRef CAS.
- A. I. Hasham, W. M. Abdel El-Azim and N. M. El-Maadawi, Egypt. J. Pet., 2007, 16, 59–80 CAS.
- T. Y. Lin, I. C. Cheng and Y. C. Hung, Appl. Phys. Lett., 2012, 101, 153701 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, DSC thermograms, compound 8 dimer in single crystal, powder X-ray diffractograms, optical images of films, CV scans of thicker films and solutions, performances of devices in a normal device configuration, and CIF for 8. CCDC 1432469. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra21681a |
| ‡ Current address: Institute of Polymer Optoelectronic Materials & Devices, South China University of Technology, Guangzhou 510640, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2016 |