
Palladium(II)-catalyzed hydroxy-involved enolate-type efficient C–C functionalization of hydroxynaphthoquinones at room temperature†
Nan Wanga,
Xingsen Wua,
Taoyu Qina,
Jianrui Zhoua,
Qidong You*a and
Xiaojin Zhang *ab
*ab
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Design and Optimization, China Pharmaceutical University, Nanjing, 210009, China. E-mail: zxj@cpu.edu.cn; youqd@163.com
bDepartment of Organic Chemistry, School of Science, China Pharmaceutical University, Nanjing, 210009, China
First published on 27th October 2016
Abstract
A simple, mild, efficient (46–92%) and scalable strategy for the synthesis of hydroxynaphthoquinones was developed through a Pd-catalyzed hydroxy-involved enolate-type C–C bond formation reaction at room temperature. The functional group tolerance exhibited by this reaction, along with the possibility of scalable production, make it a method of choice for synthesizing diverse hydroxynaphthoquinones.
Quinones are important structural units in many natural products, which exhibit redox properties that influence various regulatory cellular processes.1 Many quinone-containing compounds have biological activity owing to the presence of the quinone pharmacophore.2 Hydroxynaphthoquinones, a particular subgroup of quinones, are potent pharmaceutical substances with a well-established and wide spectrum of anti-inflammatory, anticancer, antibacterial, wound healing and antithrombotic biological activities, which contain numerous drugs and new molecular entities, such as atovaquone, buparvaquone and lapachol.3 In view of their important biological features in medicinal chemistry, a large number of hydroxynaphthoquinone derivatives and related compounds has been prepared in order to search for novel bioactive agents with improved pharmacological properties. 2-Hydroxy-1,4-naphthoquinones (HNQ; lawsone) have received particularly attention due to their biological properties and their potential as synthetic precursors of complex carbocyclic and heterocyclic quinones.3d,4 However, as shown in Fig. 1, the methods to synthesize these type of compounds is limited and low efficiency,3c,5 and in view of minimizing the side/waste product formation in 2-hydroxynaphthoquinones synthesis, the development of an additional effective synthesis protocol is necessary.
 | ||
| Fig. 1 Previous methods of synthesizing 2-hydroxynaphthoquinones.3c,5a | ||
During the past several decades, palladium-catalyzed C–C functionalization has emerged into a powerful tool for organic synthesis, which is attractive by its synthetic simplicity and step economy.6 In order to realizing atom economy and synthesis efficiency in the highly selective functionalization of C–C bond formation, much progress has been achieved so far.7 In addition, usages of various metals such as palladium(II) and copper(II)8 to promote the addition of stabilized anions onto inactivated olefins under mild reaction conditions has gained significant interest.9 More recently, the usage of a base and efficient atom-economical processes for the addition of stabilized nucleophiles onto alkenes that do not require a stoichiometric amount of transition metal have been developed.10 After examined the structure of 2-hydroxy-1,4-naphthoquinone, we reckoned that C–C bond formation at C3 position would become more easily achieved due to the existence of hydroxy at C2 position. Therefore, in this context, a Pd-catalyzed hydroxy-involved enolate-type reaction has been developed at an exceptionally mild condition into synthesising 2-hydroxynaphthoquinones.
We optimized the reaction conditions for 2-hydroxy-1,4-naphthoquinone (1a) and styrene (2a) as model substrates. When 1a (0.30 mmol) was treated with 2a (1.0 equiv. and 5.0 equiv.) in the presence of Pd(OAc)2 as catalyst, Cu(OAc)2 as oxidant and NaOAc as base in THF at 45 °C for 4 h, the desired product 3a was obtained in relatively lower yield, which is 13% and 50% (Table 1, entries 1 and 3). Initially, the reaction temperature was optimized. Yields were increased substantially by making it down to 25 °C (Table 1, entries 2 and 4). Owing to the yield differences between entries 1 and 3, we then focused on changing the equiv. of 2a (Table 1, entries 1, 4–6), and yield was up to 84% when 2a is 1.5 equiv. (Table 1, entry 5). Next followed by examining the effect of solvent. Comparing to non-polar solvent toluene, 1a were more easily deprotonated in polar solvent, such as THF, DMF, dioxane and EtOH, therefore explained the varied yields and THF remained the best solvent for the reaction among the tested solvents (Table 1, entries 5, 7–11). To be noticed, the acidic environment created by AcOH were not conducive to deprotonating process conducted by weak base as shown in entry 11. Furthermore, oxidants and catalysts were tested. Cu(OAc)2 turned to be the optimized choice of tested oxidants, which were CAN (ceric ammonium nitrate), HTIB (Koser's regent), K2S2O8, PDC (pyridinium dichromate), and KMnO4 (Table 1, entries 5, 12–16). Meanwhile the catalyst Pd(OAc)2 was replaced by PdCl2, Pd(TFA)2 and Pd(dba)2, but poor results were observed (Table 1, entries 5, 17–19). We last conducted the reaction under the nitrogen and oxygen atmosphere with oxidant Cu(OAc)2 existed. The isolated yield was decreased (Table 1, entries 5 and 20) under nitrogen atmosphere and no obvious yield increasing was observed under oxygen atmosphere (Table 1, entries 5 and 21). Therefore, the optimized reaction conditions were concluded as 2a (1.5 equiv.), Pd(OAc)2 (10 mol%), Cu(OAc)2 (1.0 equiv.), NaOAc (1.0 equiv.), and THF (solvent) at 25 °C for 4 h under air.
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2a equiv. | Temp [°C] | Oxidant | Catalyst | Solvent | Yieldb [%] |
| a Reaction condition: 1a (0.30 mmol), oxidant (1.0 equiv.), base (NaOAc, 1.0 equiv.) and Pd catalyst (10 mol%) in solvent (10 mL) for 4 h under air.b Isolated yield.c Under nitrogen.d Under oxygen. | ||||||
| 1 | 5 | 45 | Cu(OAc)2 | Pd(OAc)2 | THF | 13 |
| 2 | 5 | 25 | Cu(OAc)2 | Pd(OAc)2 | THF | 39 |
| 3 | 1 | 45 | Cu(OAc)2 | Pd(OAc)2 | THF | 50 |
| 4 | 1 | 25 | Cu(OAc)2 | Pd(OAc)2 | THF | 71 |
| 5 | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | THF | 84 |
| 6 | 3 | 25 | Cu(OAc)2 | Pd(OAc)2 | THF | 46 |
| 7 | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | DMF | 70 |
| 8 | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | EtOH | 71 |
| 9 | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | Dioxane | 44 |
| 10 | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | Toluene | 21 |
| 11 | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | AcOH | Trace |
| 12 | 1.5 | 25 | CAN | Pd(OAc)2 | THF | 63 |
| 13 | 1.5 | 25 | HTIB | Pd(OAc)2 | THF | 47 |
| 14 | 1.5 | 25 | K2S2O8 | Pd(OAc)2 | THF | 70 |
| 15 | 1.5 | 25 | PDC | Pd(OAc)2 | THF | 56 |
| 16 | 1.5 | 25 | KMnO4 | Pd(OAc)2 | THF | 71 |
| 17 | 1.5 | 25 | Cu(OAc)2 | PdCl2 | THF | 72 |
| 18 | 1.5 | 25 | Cu(OAc)2 | Pd(TFA)2 | THF | 54 |
| 19 | 1.5 | 25 | Cu(OAc)2 | Pd(dba)2 | THF | 70 |
| 20c | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | THF | 71 |
| 21d | 1.5 | 25 | Cu(OAc)2 | Pd(OAc)2 | THF | 85 |
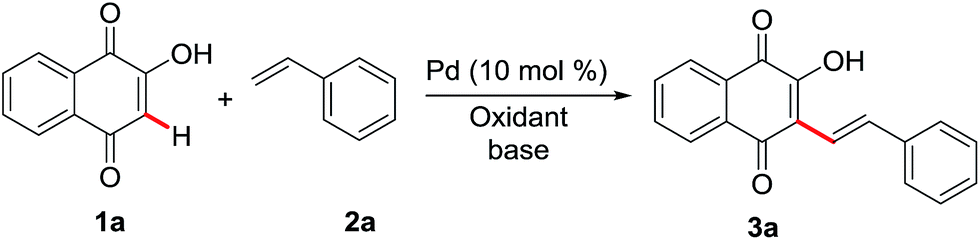
With the optimized reaction conditions, other hydroxynaphthoquinones with various side chains substituted in C3 position were systematically screened. As shown in Table 2, relatively broad range of hydroxynaphthoquinone derivatives with different substituted styrene at the C3 position of the naphthoquinone ring were successfully synthesized in good yields. The reaction outcomes were influenced by the electronic nature of substituted groups. Styrenes substituted by electron-donating groups such as –Me and –OMe achieved higher yields than by electron-withdrawing groups such as –F, –Cl. However, comparing to –Me substituted styrene (3b, 87%), –CN substituted styrene (3m) achieved the same yield, which probably caused by the conjugated effect of –CN which promoted the C–C bond formation.
|
||||
|---|---|---|---|---|
| Entry | Product | 3 | Substituted group | Yieldb [%] |
| a Reaction condition: 1a (0.30 mmol), 2 (0.45 mmol, 1.5 equiv.), Cu(OAc)2 (1.0 equiv.), NaOAc (1.0 equiv.) and Pd catalyst (10 mol%) in THF (10 mL) for 4 h under air.b Isolated yield. | ||||
| 1 |  |
3b | R3 = 4′-Me | 87 |
| 2 | 3c | R3 = 2′-Me, 4′-Me | 89 | |
| 3 | 3d | R3 = 2′-Me, 5′-Me | 88 | |
| 4 | 3e | R3 = 4′-OMe | 91 | |
| 5 | 3f | R3 = 2′-OMe | 90 | |
| 6 | 3g | R3 = 2′-OMe, 4′-OMe | 92 | |
| 7 | 3h | R3 = 4′-F | 66 | |
| 8 | 3i | R3 = 4′-Cl | 73 | |
| 9 | 3j | R3 = 2′-Cl | 74 | |
| 10 | 3k | R3 = 2′-Cl, 4′-Cl | 55 | |
| 11 | 3l | R3 = 4′-Br | 70 | |
| 12 | 3m | R3 = 4′-CN | 87 | |
| 13 | 3n | R3 = 2′-Me, 4′-F | 77 | |

Influence of different aryl ethylenes on the yield efficiency was then examined. As shown in Scheme 1, in comparison with phenyl ring (3a), replacement of thiophene ring (3p) resulted in lower yield (46%) whereas good yields were obtained in diphenyl substituted product (3o, 91%) and naphthalene ring substituted product (3q, 88%). Due to the steric effects, yield declination (67%) was observed when changed into anthracene ring (3r). Furthermore, outcomes shown in Scheme 1 revealed that changing the reacted terminal alkene from mono-substituted into double substituted decreased the yield of desired products 3s–3u down to 47–51% and a higher temperature (50 °C) was required.
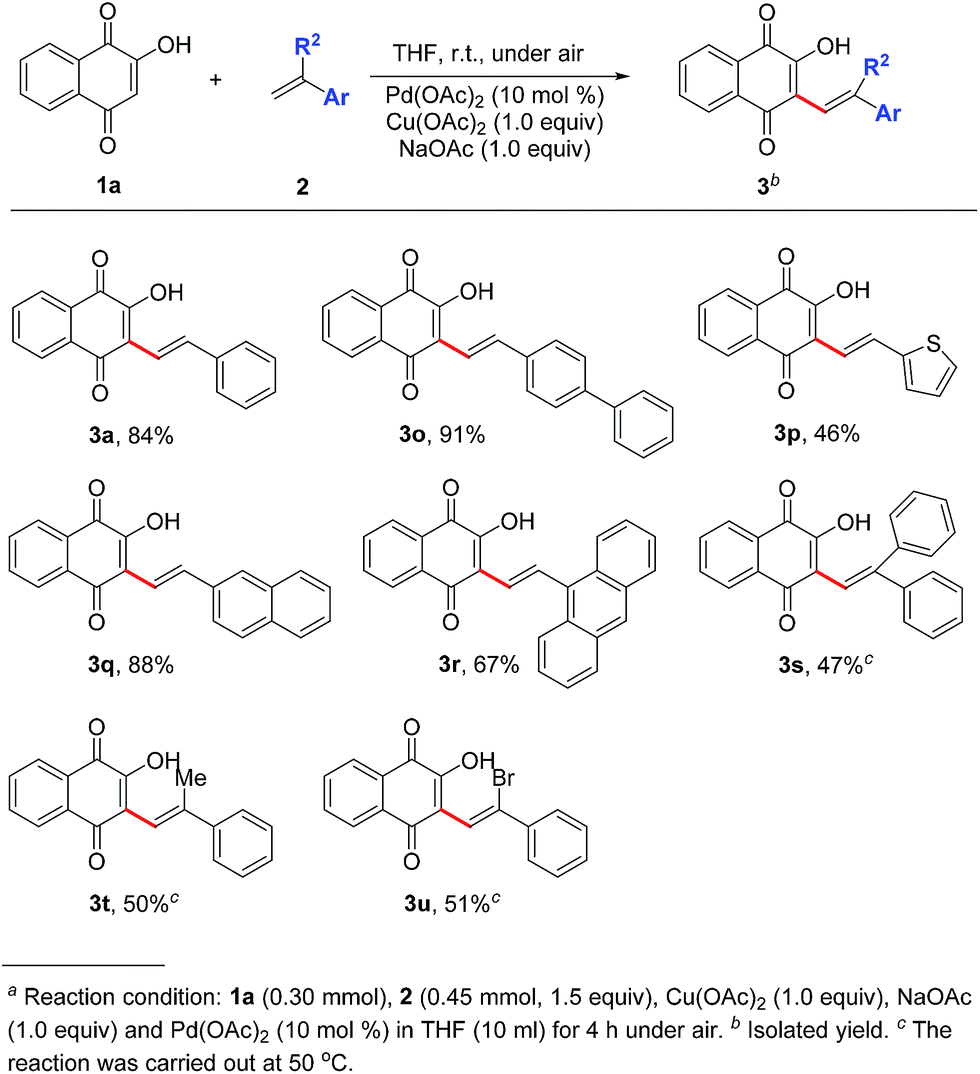 | ||
| Scheme 1 Substrate scope of vinyl-substituted aromatic hydrocarbons and ipsilateral double substituteda ethylenes. | ||
To further expand the reaction usability and substrate scope, our attention was next turned towards testing the feasibility of replacing 1a in C5, C6 and C7 positions and matching them with different 2 scoped previously. Those trying of combinations reacted well and achieved the corresponding products 3v–3af in 82–90% yield (Table 3).
|
||||
|---|---|---|---|---|
| Entry | Product | 3 | Substituted group | Yieldb [%] |
| a Reaction condition: 1a (0.30 mmol), 2 (0.45 mmol, 1.5 equiv.), Cu(OAc)2 (1.0 equiv.), NaOAc (1.0 equiv.) and Pd catalyst (10 mol%) in THF (10 mL) for 4 h under air.b Isolated yield. | ||||
| 1 |  |
3v | R1 = 7-Me | 86 |
| 2 | 3w | R1 = 7-Me; R3 = 4′-Cl | 84 | |
| 3 | 3x | R1 = 7-Me; R3 = 4′-Br | 83 | |
| 4 | 3y | R1 = 5-OMe | 83 | |
| 5 | 3z | R1 = 7-OMe | 86 | |
| 6 | 3aa | R1 = 7-OMe; R3 = 4′-Me | 89 | |
| 7 | 3ab | R1 = 6,7-OMe | 90 | |
| 8 | 3ac | R1 = 7-Cl | 84 | |
| 9 | 3ad | R1 = 7-Cl; R3 = 4′-Me | 85 | |
| 10 | 3ae | R1 = 7-Cl; R3 = 4′-Br | 82 | |
| 11 | 3af | R1 = 7-Br | 83 | |

To demonstrate the practicability of the method, the model procedure was successfully scaled up with comparable yield. The desired product 3a (3.19 g, 77%) was prepared when the reaction was run in 15 mmol scale (Scheme 2). The recycling of the catalyst is an important issues in reactions using heterogeneous catalysis.11 When scaled up to this amount, the heterogeneous catalysis used in the reaction can be recycled through filtration and drying overnight and the yield of next cycle was acceptable (72%).
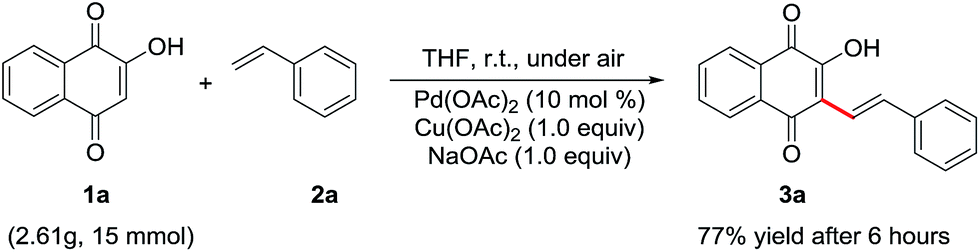 | ||
| Scheme 2 Scale-up experiment. | ||
To gain insight into the reaction mechanism, several additional control experiments (Scheme 3) were conducted. When replacing –OH of 1a in C2 position into –OMe and –H and performing the reaction with substrate 2a in standard condition, no reaction was observed. Based on these two control experiments, it is speculated that differentiated with general C–C bond formation, –OH in C2 position participated in the reaction and played the crucial role of carrying on the integrate mechanism.
 | ||
| Scheme 3 Control experiments. | ||
Employing model substrates 1a and 2a for illustrative purposes, a possible catalytic mechanism cycle for these reactions is shown in Scheme 4. After the first step forming the complex A as a sodium salt, exchanging between the palladium acetate and the alkoxide gives the complex B, which is the η-1 tautomer of η-3 palladium enolate C. Then C interconverts to the other tautomer, η-1 palladium enolate D and further followed by the migratory alkene insertion, which forms complex E. The obtained E undergoes β-H elimination and tautomerization to restore the aromaticity and therefore achieve the goal product 3a and Pd(0) species subsequently. At last, Pd(0) catalyst is reoxidized by 1.0 equiv. of Cu(OAc)2 and regenerates the Pd(II) catalyst as a result which completed the catalytic cycle.
 | ||
| Scheme 4 Possible mechanism for reaction. | ||
Conclusions
We have developed a simple, mild, high-yield and scalable strategy for the synthesis of hydroxynaphthoquinones via a Pd-catalyzed hydroxy-involved enolate-type reaction at room temperature. Comparing to various reported Pd(II)-catalyst system, the mechanism of this synthetic strategy involved Pd-enolate intermediate transferred by weak base NaOAc so that the reaction can be conducted in a mild condition and the recovery of catalyst system made it practically for further industrial scale-up operation. Additionally, how electronic nature, aromaticity and steric factor have influenced the reaction yields was discussed and the mechanisms of this reaction were brought up with based on the 2-position hydroxy involved C–C functionalization. Functional group tolerance exhibited by this reaction along with the possibility of scalable production make it a method of choice of synthesizing hydroxynaphthoquinones. Further investigations of this reaction for the mechanism and its applications are currently underway in our laboratory.Experimental section
Representative procedure for the synthesis of 3a
A mixture of 2-hydroxy-1,4-naphthoquinone (1a, 0.30 mmol, 53 mg, 1.0 equiv.), styrene (2a, 0.45 mmol, 47 mg/0.051 mL, 1.5 equiv.), Cu(OAc)2 (0.30 mmol, 54 mg, 1.0 equiv.), NaOAc (0.30 mmol, 41 mg, 1.0 equiv.), Pd(OAc)2 (6.7 mg, 10 mol%) and THF (solvent, 10 mL) was stirred for 4 h at 25 °C under air condition. Upon completion of the reaction, as determined by TLC, the mixture was filtered through a celite pad, and then washed with cooled water (20 mL), extracted with dichloromethane (3 × 20 mL) and dried over anhydrous Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel using dichloromethane as the eluent to give 3a as a red powder; yield: 70 mg (84%): mp 145.2–146.8 °C.Acknowledgements
We are thankful for the financial support of the National Natural Science Foundation of China (No. 81302636, 81230078, 81573346, 81603025), the Natural Science Foundation of Jiangsu Province (No. BK20130656), the National Major Science and Technology Project of China (No. 2013ZX09402102-001-005, 2014ZX09507002-005-015), the Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP, 20130096110002), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- (a) W. Yang and D.-M. Du, Adv. Synth. Catal., 2011, 353, 1241–1246 CrossRef CAS; (b) A. N. Assimopoulou and V. P. Papageorgiou, Phytother. Res., 2005, 19, 141–147 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Glänzel, R. Bültmann, K. Starke and A. W. Frahm, Eur. J. Med. Chem., 2005, 40, 1262 CrossRef PubMed; (b) V. K. Tandon, R. B. Chhor, R. V. Singh, S. Rai and D. B. Yadav, Bioorg. Med. Chem. Lett., 2004, 14, 1079 CrossRef CAS PubMed; (c) V. F. de Andrade-Neto, M. O. F. Goulart, J. F. da Silva Filho, M. J. da Silva, M. C. F. R. Pinto, A. V. Pinto, M. G. Zalis, L. H. Carvalho and A. U. Krettli, Bioorg. Med. Chem. Lett., 2004, 14, 1145 CrossRef CAS PubMed; (d) I. Gomez-Monterrey, G. Santelli, P. Campiglia, D. Califano, F. Falasconi, C. Pisano, L. Vesci, T. Lama, P. Grieco and E. Novellino, J. Med. Chem., 2005, 48, 1152 CrossRef CAS PubMed.
- For selected examples, see: (a) F. Zsila and I. Fitos, Org. Biomol. Chem., 2010, 8, 4905–4914 RSC; (b) S. Shaabani, M. R. Naimi-Jamal and A. Maleki, Dyes Pigm., 2015, 122, 46–49 CrossRef CAS; (c) S. Fiorito, F. Epifano, C. Bruyere, V. Mathieu, R. Kiss and S. Genovese, Bioorg. Med. Chem. Lett., 2014, 24, 454–457 CrossRef CAS PubMed; (d) J. R. Dai, L. A. Decosterd, K. R. Gustafson, J. H. Cardellina II, G. N. Gray and M. R. Boyd, J. Nat. Prod., 1994, 57, 1511–1516 CrossRef CAS PubMed.
- T. Ogata, T. Yoshida, M. Shimizu, M. Tanaka, C. Fukuhara, J. Ishii, A. Nishiuchi, K. Inamoto and T. Kimachi, Tetrahedron, 2016, 72, 1423–1432 CrossRef CAS.
- (a) E. Malamidou-Xenikaki, M. Tsanakopoulou, M. Chatzistefanou and D. H. Litina, Tetrahedron, 2015, 71, 5650–5661 CrossRef CAS; (b) J.-L. Bian, X. Qian, N. Wang, T. Mu, X. Li, H.-P. Sun, L. Zhang, Q.-D. You and X.-J. Zhang, Org. Lett., 2015, 17, 3410–3413 CrossRef CAS PubMed; (c) K. H. Lee and H. W. Moore, Tetrahedron Lett., 1993, 34, 235–238 CrossRef CAS.
- (a) S. E. Walker, J. A. Jordan-Hore, D. G. Johnson, S. A. Macgregor and A. L. Lee, Angew. Chem., Int. Ed., 2014, 53, 13876–13879 CrossRef CAS PubMed; (b) M. T. Molina, C. Navarro, A. Moreno and A. G. Csákÿ, Org. Lett., 2009, 11, 3928–4941 CrossRef PubMed; (c) S. Cai, C. Chen, Z. Sun and C. Xi, Chem. Commun., 2013, 49, 4552–4554 RSC; (d) R. Giri, S. Thapa and A. Kafle, Adv. Synth. Catal., 2014, 356, 1395–1411 CrossRef CAS; (e) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed; (f) C. Röhlich and K. Köhler, Adv. Synth. Catal., 2010, 352, 2263–2274 CrossRef.
- (a) M.-F. Zheng, L.-B. Huang, W.-Q. Wu and H.-f. Jiang, Org. Lett., 2013, 15, 1838–1841 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (c) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS; (d) E. M. Beccalli, G. Broggini, S. Gazzola and A. Mazza, Org. Biomol. Chem., 2014, 12, 6767–6789 RSC.
- Y. Li, Z.-K. Yu and S.-Z. Wu, J. Org. Chem., 2008, 73, 5647–5650 CrossRef CAS PubMed.
- (a) F. Dénès, A. Pérez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366–2447 CrossRef PubMed; (b) G. Balme, E. Bossharth and N. Monteiro, Eur. J. Org. Chem., 2003, 24, 4101–4111 CrossRef.
- For selected examples, see: (a) A. G. Fallis and P. Forgione, Tetrahedron, 2001, 57, 5899–5913 CrossRef CAS; (b) J. V. N. Vara Prasad and C. N. Pillai, J. Organomet. Chem., 1983, 259, 1–4 CrossRef; (c) E.-i. Negishi, Pure Appl. Chem., 1981, 53, 2333–2356 CrossRef CAS; (d) M. Yamaguchi and Y. Nishimura, Chem. Commun., 2008, 35–48 RSC; (e) A. Pérez-Luna, C. Botuha, F. Ferreira and F. Chemla, New J. Chem., 2008, 32, 594–606 RSC.
- A. Modak, J. Mondal, V. k. Aswal and A. Bhaumik, J. Mater. Chem., 2010, 20, 8099–8106 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21652h |
| This journal is © The Royal Society of Chemistry 2016 |