
Size and pH dependent photoluminescence of graphene quantum dots with low oxygen content†
Sung-Ho Song a,
Minho Jangb,
Hyewon Yoonc,
Yong-Hoon Chob,
Seokwoo Jeon*c and
Bo-Hyun Kim*d
a,
Minho Jangb,
Hyewon Yoonc,
Yong-Hoon Chob,
Seokwoo Jeon*c and
Bo-Hyun Kim*d
aDivision of Advanced Materials Engineering, Kongju National University, Chungnam 330-717, Korea
bDepartment of Physics, KAIST, Daejeon 34141, Korea
cDepartment of Materials Science and Engineering, Graphene Research Center of KI for the NanoCentury, KAIST, Daejeon 34141, Korea. E-mail: jeon39@kaist.ac.kr
dSchool of Basic Science, Convergence College, DGIST, Daegu 42988, Korea. E-mail: bhkim@dgist.ac.kr
First published on 6th October 2016
Abstract
The photoluminescence of graphene quantum dots (GQDs) is rigorously investigated due to their potential applications. However, GQDs from graphene oxide are inherently embedded with non-negligible defects and oxygen grafted onto the edge and basal plane, which induce a change of innate electronic structure in the GQDs. Thus, graphene oxide based GQDs can misrepresent the characteristic properties of primitive GQDs. Here we report the size and pH dependent photophysical properties of GQDs that minimize the content of oxygen and defects. From auger electron spectroscopy, the oxygen content of the GQDs with two different lateral sizes (∼2 nm and ∼18 nm) was probed and found to be ∼5% and ∼8%, respectively. Two common photoluminescence (PL) peaks were observed at 436 nm (the intrinsic bandgap) and 487 nm (the extrinsic bandgap) for both GQDs. The characteristic PL properties in extrinsic and intrinsic bandgaps examined by optical spectroscopic methods show that the emission peak was red-shifted and that the peak width was widened as the size increased. Moreover, the PL lifetime and intensity were not only reversibly changed by pH but also depended on the excitation wavelength. This is in line with our previous report and is ascribed to the size variation of sp2 subdomains and edge functionalization.
1. Introduction
Graphene quantum dots (GQDs) have recently attracted a great deal of interest because of their chemical inertness, low cytotoxicity and strong photoluminescence (PL).1 They are presently being rigorously investigated for a range of applications in electronics, optoelectronics, medicine, bioimaging, and energy conversion systems.1–6 In particular, the GQDs’ widely tunable bandgap enables them to be utilized as a potential alternative to inorganic quantum dots.7 However, their electronic and optical properties are strongly affected by the quality of the GQDs, which depends on the fabrication process.7–12 Defects and oxygen content are critical factors which can influence the electronic energy structure of GQDs. The origin of the GQDs’ photoluminescence has been widely discussed, and it is believed that isolated subdomains of sp2 carbon, mainly composed of four to seven carbon hexagons, are the origin of the intrinsic luminescence of GQDs.3,9,13 However, further studies are still needed to determine the photophysical properties of GQDs under specific conditions, such as at different sizes and pH.The important parameters affecting the photophysical properties of GQDs are their size dependent quantum confinement, their edge structure and functionalization, and their exciton binding energy.7,8,14–17 Ritter et al. and Lu et al. showed that the band gap of GQDs increased as their size decreased, and that the edge structure of the GQDs influenced their electronic structure.18,19 Blue-shifted emissions were observed when their size was decreased.20 Jin et al. reported a PL shift resulting from charge transfer between amine functional groups and the graphene oxide (GO) based GQDs.7 Recently, Kumar et al. asserted that the interband states induced by orbital hybridization between C and N atoms led to multicolor emission with a change in excitation wavelength.21 Up to now, the most plausible emission mechanism for GQDs has been exciton decay from bands which have been classified as intrinsic and extrinsic.3,22,23 It has been suggested that the intrinsic band, which mainly emits a blue color, originates in isolated sp2 bonded carbon domains. Huang et al. theoretically discussed the anomalous and wide photoluminescence of GQDs, and suggested that the edge localized exciton of GQDs exhibits size- and pH-dependent absorption/emission.17 Recently, Yoon et al. demonstrated that the bright blue emission of GQDs was due to the favorable formation of subdomains composed of four to seven carbon hexagons.9 Meanwhile, the extrinsic band, which corresponds to red colored emissions, is attributed to oxidative defects and edge-based functional groups.
For years, various methods and techniques have been introduced for preparing high quality GQDs.3,9,10,12,19,24 However, harsh chemicals are used in most GQD fabrication processes, and in GO based GQDs this inevitably results in defects and oxygen containing functional groups like hydroxyl, carboxyl and epoxy groups.7,18,25,26 To address this issue, we have developed a series of fabrication methods for high quality graphene, graphene flakes and GQDs.3,7,11,12,27 Most recently, we introduced a modified graphite intercalated compounds (GICs) method, which enabled us to mass produce GQDs under relatively less harsh conditions and at a lower temperature.3 Based on this method, we have investigated the origin of the blue emissions in GQDs, which has been attributed to the formation of subdomains.9
In this study, we investigated the effect of size and pH on the photophysical properties of the GQDs prepared by a modified GICs method. We prepared two sizes of GQDs (2 nm and 18 nm) and their oxygen content was ∼5 at% and ∼8 at%, respectively, as probed by auger electron spectroscopy. The size and pH dependent PL properties of the GQDs were analyzed based on a model with intrinsic and extrinsic electronic states. The emission decay from the intrinsic band (∼420 nm) or the extrinsic band (∼490 nm) depended on their size and the excitation wavelength. pH had the opposite effect on GQD-A and -B. However, the PL properties of the GQDs were reversible and reproducible by repeated protonation and deprotonation.
2. Experimental
Graphene quantum dots (GQDs)
Fig. S1† schematically shows the fabrication process of GQDs through the GICs method. For the preparation of GQDs from GICs, 20 mg of graphite powder and 300 mg of potassium–sodium tartrate tetrahydrate (KNaC4H4O6·4H2O) powder were mixed using a pestle and then heated to 250 °C for 24 h under an Ar atmosphere. The heated mixture was then cooled down, and 20 ml of water was added. Next, the solution was sonicated for 24 h and dialyzed by a 2 kDa dialysis pack, finally obtaining a high quality GQD solution. All of the samples were microcentrifuged with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MWCO and 30000 MWCO microfilters for size separation. More details were reported in a previous report.3
000 MWCO and 30000 MWCO microfilters for size separation. More details were reported in a previous report.3
Structural characterization
The morphology of the GQDs was analyzed using transmission electron microscopy (TEM, Tecnai G2 F30) and atomic force microscopy (AFM, SPA400, SII, Japan) in tapping mode under ambient conditions. TEM samples were prepared by drying a droplet of the GQD suspension on a carbon grid. Raman spectra were obtained between 1200 to 3000 cm−1 using a Raman spectrometer (LabRAM HR UV/Vis/NIR, excitation at 514 nm). UV/Vis spectra (UV-3101 PC spectrometer) and FT-IR spectra (FT-IR-4100 type-A FT-IR spectrometer) were obtained. AES analysis was conducted with a source electron beam energy of 5 kV (Perkin Elmer). Solution phase photoluminescence (PL) spectra were obtained using a quartz cuvette under an excitation wavelength of 325 nm from monochromatic light formed by a 150 W Xenon lamp at room temperature (deionized water was used as the solvent).Photoluminescence characterization
All the luminescence data were obtained by a handmade setup using precision cells made of quartz suprasil. The photoluminescence (PL) measurements, such as pH-dependent, size-dependent, and excitation wavelength-dependent PL behaviors, were taken using a 325 nm He–Cd continuous-wave (CW) laser, monochromatic light from a 300 W-xenon lamp, and a UV spectrometer (Maya2000, Ocean Optics, USA) as a PL detector at room temperature. The PL excitation was measured by monochromatic light from a 300 W Xenon lamp and a high-sensitivity photomultiplier tube as a PL detector. A bandgap-locked femto-second pulsed Ti:sapphire laser (Coherent, Chameleon Ultra II) system was used as an excitation source, and five wavelengths of the pulsed Ti:sapphire laser (266 nm, 300 nm, 350 nm, 400 nm, and 450 nm) were employed.3. Results and discussion
After the synthesis of the GQDs, two different sizes of GQDs were selectively prepared using a molecular weight cut-off membrane (10000 Da. and 30000 Da). Fig. 1a and b are TEM images of the GQDs centrifuged with a 10000 Da membrane (GQD-A) and a 30000 Da membrane (GQD-B), respectively. The crystalline structure of the GQDs was confirmed in the previous report.3 From the histograms of the statistical analysis, GQD-A and -B were estimated to have uniform sizes of 2 ± 1 nm and 18 ± 2 nm, respectively. Most of the GQDs were estimated to have about 3 layers or less from the height profile obtained from the AFM images (Fig. S2†). In the FT-IR spectra of GQD-A and -B (Fig. 1c), the intensity of the –OH vibration peak was relatively lower than other characteristic peaks, suggesting that the total oxygen content of GQD-A and -B was generally less than that of GO based GQDs, or even reduced GO based GQDs.3,7,28,29 Comparing GQD-A and -B, the oxygen content of GQD-A was lower than that of GQD-B. Among the characteristic peaks, the carboxylic acid related peaks (ca. 1720 cm−1 and ca. 1225 cm−1) in GQD-A disappeared or were dramatically decreased compared to those of GQD-B. Although the peak at ca. 1225 cm−1 originating from epoxy groups or carboxyl groups was observed in both GQDs, other characteristic absorption peaks for epoxy (ca. 854 cm−1 and 970 cm−1) were not observed. This means that most of the oxygen related peaks came from the edge functionalized carboxyl groups, supporting the theory that our GQDs have few defects on the basal plane. Raman spectra (Fig. S3†) also suggested that GQD-A had more robust carbon sp2 bonding and fewer defects compared to GQD-B. In the UV-Vis absorption spectra obtained from GQD-A and -B, the absorption peak which relates to the n–π* transition (ca. 350 nm) was nearly suppressed in GQD-A but was still present in GQD-B (Fig. S4†). This means that orbital hybridization due to O–C bonding was less formed in GQD-A. For a quantitative analysis of the chemical elements in the GQDs, we adopted the auger electron spectroscopy (AES) method. From the AES spectra of GQD-A and -B (Fig. 1d), the oxygen content was estimated to be 5 at% and 8.3 at%, respectively, which is in accordance with our previous results.3,9
 | ||
Fig. 1 Characterization of the GQDs; (a and b) HRTEM images and fringe patterns of GQD-A and GQD-B, respectively, insets show the size distribution in GQD-A and GQD-B, respectively. (c) FT-IR spectra of GQD-A and GQD-B. Characteristic peaks shown; the hydroxyl (–OH) band, the C–H band (1524 cm−1), the carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–) or carboxyl (–COOH) band (1720 cm−1), and the –OH band (1379 cm−1). (d) AES spectra of GQD-A and GQD-B on a Si/SiO2 substrate at a source electron beam energy of 5 kV, respectively. O–) or carboxyl (–COOH) band (1720 cm−1), and the –OH band (1379 cm−1). (d) AES spectra of GQD-A and GQD-B on a Si/SiO2 substrate at a source electron beam energy of 5 kV, respectively. | ||
The PL properties of GQD-A and -B were investigated under varying excitation wavelengths (λex). Fig. 2a shows the PL spectra of GQD-A and -B excited by λex = 310 nm. We undertook spectrum deconvolution into two sub peaks with a PL emission model on the basis of intrinsic and extrinsic electronic bands. The shorter emission wavelengths (λem ∼ 419 nm in GQD-A and 430 nm in GQD-B) were assigned to emission decay from the intrinsic band, and the longer emission wavelengths (492 nm in GQD-A and 510 nm in GQD-B) to the extrinsic band, as illustrated in the inset of Fig. 2a. Since the extrinsic band originates from defects and oxygen functional groups, its peak center is red-shifted from that of the intrinsic emission decay. In GQD-A, the integrated intensity ratio between the deconvoluted peaks was 0.63, whereas in GQD-B it was 0.46. This implies that the intrinsic bands in GQD-A are mainly used for emission decay pathways, whereas in GQD-B the extrinsic bands are dominantly used for charge transfer.3,13 As the GQD size increased the emission peaks were red-shifted (λem: 419 nm → 430 nm, 492 nm → 510 nm) and broadened up to 120%. This might reflect the size effect of the aromatic sp2 nano-domains and the band-edge overlap between the intrinsic and extrinsic bands due to the increase in defects and functional groups.3,9,13 Fig. 2b and c show the λex dependent PL (PLE) measured at the center of the peaks of the intrinsic and extrinsic PL. In GQD-A (Fig. 2b), λex ∼ 320 nm resulted in a higher intensity for the intrinsic PL (λem = 419 nm) and a similar intensity for the extrinsic PL (λem = 492 nm) compared to the longer excitation wavelength (λex ∼ 355 nm). This indicates that charge transfer from the intrinsic band into the extrinsic band occurred briskly. At the same time, in GQD-B the λex ∼ 360 nm predominantly affected the extrinsic PL intensity (Fig. 2c). This suggests that as the size increased, the density of the extrinsic states was increased and direct emission decay from them became dominant. This is in line with the above results from Raman spectroscopy and AES, which indicated that the defect and oxygen content increased as the size of the GQDs increased. Defects and oxygen functional groups provide extrinsic electronic states which can induce PL and reduce the lifetimes of GQDs.
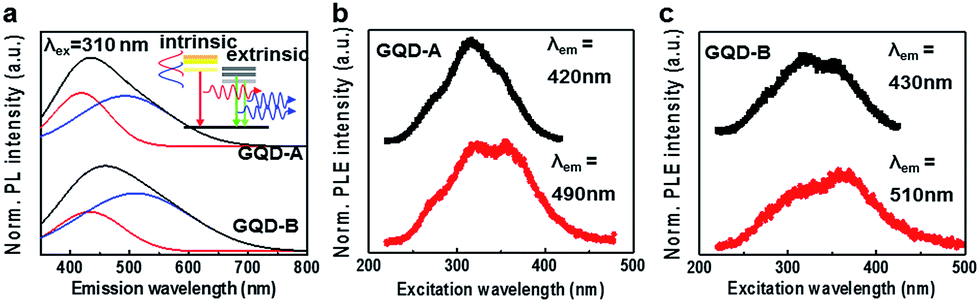 | ||
| Fig. 2 Optical characterization of GQD-A and GQD-B (a). PL spectra of GQD-A and GQD-B under excitation at 310 nm of monochromatic light from a xenon lamp. The PL spectra of GQD-A and GQD-B show blue and green emission due to the oxygen content. (b and c) PLE spectra of GQD-A and GQD-B. The PLE spectra were measured at the maximum PL peak position of both GQD-A and GQD-B. | ||
Song et al. demonstrated that as the size of GQDs increased the PL intensity was decreased.3 To address the issue of PL lifetime, we measured the time resolved PL (TRPL) decay profile with three different excitation wavelengths; λex = 266 nm for intrinsic states, λex = 355 nm for intrinsic and extrinsic states, and λex = 400 nm for the extrinsic bandgap. Fig. 3a and b show the intensity integrated TRPL decay profiles measured for GQD-A and GQD-B, respectively.
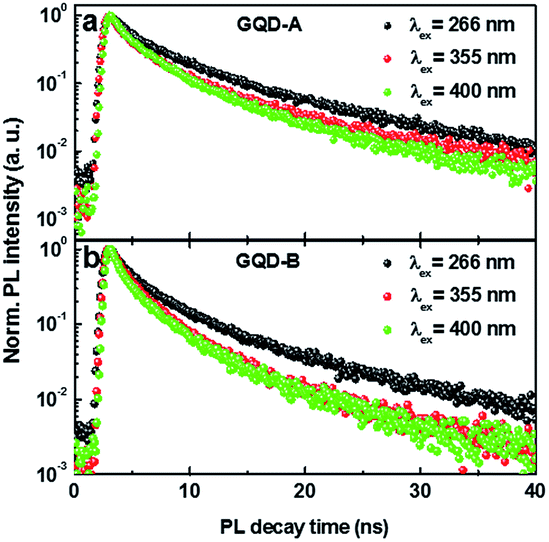 | ||
| Fig. 3 TRPL of GQD-A and GQD-B under different excitation wavelengths. (a) TRPL of GQD-A under excitation with wavelengths of 266 nm, 355 nm and 400 nm. (b) TRPL of GQD-B with the same excitation wavelengths as those used on GQD-A. | ||
The PL decay lifetimes were estimated from the fitting curve using a double exponential decay function with minimum reduced chi-squared value. The estimated t1 and t2 values are listed in Table 1. t1 is related to the emission decay from the intrinsic states while t2 is from the extrinsic states. As the λex increased from 266 nm to 400 nm, the lifetimes of both sizes of GQDs were decreased by more than 30%. The longest t2 at λex = 266 nm is attributed to charge transfer from the intrinsic state to the extrinsic state. On the other hand, the shortest value of t2 at λex = 400 nm is most likely due to the direct excitation and decay of charges from the extrinsic states. Although t1 and t2 show a similar trend in both GQD-A and -B, they were relatively shorter in GQD-B, supporting the conclusion that in the bigger GQD-B there are more PL quenching sites originating from defects and oxygen functional groups. This is confirmed by the result that the smaller GQDs show a higher quantum yield of PL.3
| λex | 266 nm | 355 nm | 400 nm | |||
|---|---|---|---|---|---|---|
| Lifetime | t1 | t2 | t1 | t2 | t1 | t2 |
| GQD-A | 1.84 ± 0.04 | 7.89 ± 0.16 | 1.25 ± 0.02 | 5.97 ± 0.10 | 1.15 ± 0.02 | 5.06 ± 0.08 |
| GQD-B | 1.27 ± 0.02 | 6.11 ± 0.09 | 1.09 ± 0.02 | 4.30 ± 0.07 | 0.84 ± 0.01 | 4.03 ± 0.05 |
Fig. 4a and b show a comparison of the PL from protonated (pH 2) and deprotonated (pH 12) GQD-A and GQD-B, respectively. All the PL spectra are deconvoluted into intrinsic and extrinsic emission modes similar to the analysis in Fig. 2a. When GQD-A was repeatedly protonated and deprotonated, the integrated PL intensity was not significantly changed, although it showed a sharpened PL peak under protonation conditions.
 | ||
| Fig. 4 Comparison of the optical properties of GQD-A and GQD-B with size dependence; (a) pH-dependent PL of GQD-A and (b) pH-dependent PL of GQD-B. The PL was carried out under reversible pH-solvent conditions. (c) Emission images of GQD-B dispersed in water under reversible pH-solvent conditions. | ||
However, in GQD-B, although the center of the deconvoluted PL peaks appeared at a similar wavelength, the integrated area ratio of the intrinsic mode was increased by about 25% when GQD-B was protonated, compared to the ratio under deprotonation conditions. This implies that the (de)protonation of GQD-A didn’t critically affect the PL properties. Meanwhile, the (de)protonation of GQD-B significantly changed the PL properties. Fig. 4c photographically illustrates the dramatic change in the PL of GQD-B with changing pH. We attribute this to the difference in defect and oxygen content between GQD-A and -B. This is in accordance with the above discussed results and previous reports.3,9,13
The PL of the GQDs was measured in three different pH solutions with varying excitation wavelengths. Fig. 5a–c show the PL of GQD-A measured at pH 2, 7 and 12, respectively. For GQD-A, at all pH values the highest PL intensity was observed when excited at λex = 315 nm. Farther from the resonance excitation wavelength and with increasing pH, the PL intensity is decreased. When protonated, the PL profiles of GQD-A were sharper than those at other pH values. In addition, the center of the emission profiles at pH 2 was blue-shifted compared to those at pH 7 and 12. This might be due to the resonance excitation of the intrinsic band and a protonation effect on the edge and defect sites. One thing we noticed is that the PL intensity was sharply increased at λex = 360 nm, which is due to the resonance excitation of the extrinsic band. The PL intensity at λex = 315 nm was gradually decreased as pH increased, while that at λex = 360 nm slightly fluctuated. This might depend on the efficiency of the charge transfer and the density of the extrinsic states. However, GQD-B showed the opposite trend in PLE properties according to the pH (Fig. 5d–f). At pH 2 (Fig. 5d), GQD-B shows the lowest PLE intensity. As the pH was increased the intensity increased. As expected, at pH 7 (Fig. 5e) and pH 12 (Fig. 5f) the PL intensity at λex = 360 nm showed the maximum value, which is in contrast to the value at λex = 330 nm at pH 2. This can be attributed to the piling up of the extrinsic state through the deprotonated defects and edges.
 | ||
| Fig. 5 pH dependent PL of GQD-A and GQD-B under different excitation wavelengths. PL spectra showing the dependence on varying excitation wavelength from 250 nm to 390 nm of GQD-A at pH 2 (a), pH 7 (b) and pH 12 (c), and those of GQD-B at pH 2 (d), pH 7 (e) and pH 12 (f). | ||
4. Conclusions
So far, we have investigated the pH dependent PL properties of GQDs with two different sizes. The PL properties of GQDs were analyzed by a model based on the intrinsic and extrinsic electronic bands. GQDs with a size of ∼2 nm show less sensitivity to pH variation than those with a size of ∼18 nm. This is due to the formation of extrinsic bands originating from defects and oxygen functional groups. Our results suggest that for effective blue emission materials the smaller sized GQDs could be considered, with large advantages over the bigger sized GQDs. However, the bigger sized GQDs could be used for electroluminescent materials, with variable emission wavelengths, or as pH sensors due to their varying emission wavelengths and reversible PL properties through protonation and deprotonation. Although recently the origin of PL in GQDs has been successfully demonstrated by Yoon et al., our results show that the PL properties of GQDs strongly depend on the size and pH.Acknowledgements
This work was supported by the Center for Advanced Soft-Electronics funded by the Ministry of Science, ICT and Future Planning as Global Frontier (NRF-2013M3A6A5073173), and partially supported by the nanomaterial technology development program through the national research foundation of Korea (NRF) funded by the ministry of science, ICT and Future planning (2012M3A7B4049807).Notes and references
- G. Konstantatos, M. Badioli, L. Gaudreau, J. Osmond, M. Bernechea, F. P. G. de Arquer, F. Gatti and F. H. L. Koppens, Nat. Nanotechnol., 2012, 7, 363–368 CrossRef CAS PubMed.
- S. H. Song, M. H. Jang, J. M. Jeong, H. Yoon, Y. H. Cho, W. I. Jeong, B. H. Kim and S. Jeon, Chem. Commun., 2015, 51, 8041–8043 RSC.
- S. H. Song, M. H. Jang, J. Chung, S. H. Jin, B. H. Kim, S. H. Hur, S. Yoo, Y. H. Cho and S. Jeon, Adv. Opt. Mater., 2014, 2, 1016–1023 CrossRef CAS.
- H. J. Sun, L. Wu, W. L. Wei and X. G. Qu, Mater. Today, 2013, 16, 433–442 CrossRef CAS.
- L. B. Tang, R. B. Ji, X. K. Cao, J. Y. Lin, H. X. Jiang, X. M. Li, K. S. Teng, C. M. Luk, S. J. Zeng, J. H. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed.
- J. H. Shen, Y. H. Zhu, X. L. Yang and C. Z. Li, Chem. Commun., 2012, 48, 3686–3699 RSC.
- S. H. Jin, H. Kim da, G. H. Jun, S. H. Hong and S. Jeon, ACS Nano, 2013, 7, 1239–1245 CrossRef CAS PubMed.
- M. A. Sk, A. Ananthanarayanan, L. Huang, K. H. Lim and P. Chen, J. Mater. Chem. C, 2014, 2, 6954–6960 RSC.
- H. Yoon, Y. H. Chang, S. H. Song, E. S. Lee, S. H. Jin, C. Park, J. Lee, B. H. Kim, H. J. Kang, Y. H. Kim and S. Jeon, Adv. Mater., 2016, 26, 5255–5261 CrossRef PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. H. Ge, L. Song, L. B. Alemany, X. B. Zhan, G. H. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J. J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- K. H. Park, B. H. Kim, S. H. Song, J. Kwon, B. S. Kong, K. Kang and S. Jeon, Nano Lett., 2012, 12, 2871–2876 CrossRef CAS PubMed.
- J. Lee, K. Kim, W. I. Park, B. H. Kim, J. H. Park, T. H. Kim, S. Bong, C. H. Kim, G. Chae, M. Jun, Y. Hwang, Y. S. Jung and S. Jeon, Nano Lett., 2012, 12, 6078–6083 CrossRef CAS PubMed.
- M. H. Jang, H. D. Ha, E. S. Lee, F. Liu, Y. H. Kim, T. S. Seo and Y. H. Cho, Small, 2015, 11, 3773–3781 CrossRef CAS PubMed.
- S. J. Zhu, J. H. Zhang, S. J. Tang, C. Y. Qiao, L. Wang, H. Y. Wang, X. Liu, B. Li, Y. F. Li, W. L. Yu, X. F. Wang, H. C. Sun and B. Yang, Adv. Funct. Mater., 2012, 22, 4732–4740 CrossRef CAS.
- S. Kim, S. W. Hwang, M. K. Kim, D. Y. Shin, D. H. Shin, C. O. Kim, S. B. Yang, J. H. Park, E. Hwang, S. H. Choi, G. Ko, S. Sim, C. Sone, H. J. Choi, S. Bae and B. H. Hong, ACS Nano, 2012, 6, 8203–8208 CrossRef CAS PubMed.
- G. D. Scholes and G. Rumbles, Nat. Mater., 2006, 5, 920 CrossRef CAS.
- P. Huang, J. J. Shi, M. Zhang, X. H. Jiang, H. X. Zhong, Y. M. Ding, X. Cao, M. Wu and J. Lu, J. Phys. Chem. Lett., 2016, 7, 2888–2892 CrossRef CAS PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS PubMed.
- J. Lu, P. S. E. Yeo, C. K. Gan, P. Wu and K. P. Loh, Nat. Nanotechnol., 2011, 6, 247–252 CrossRef CAS PubMed.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. S. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- G. S. Kumar, R. Roy, D. Sen, U. K. Ghorai, R. Thapa, N. Mazumder, S. Saha and K. K. Chattopadhyay, Nanoscale, 2014, 6, 3384–3391 RSC.
- F. Liu, M. H. Jang, H. D. Ha, J. H. Kim, Y. H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657–3662 CrossRef CAS PubMed.
- L. Wang, S. J. Zhu, H. Y. Wang, S. N. Qu, Y. L. Zhang, J. H. Zhang, Q. D. Chen, H. L. Xu, W. Han, B. Yang and H. B. Sun, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- Y. Q. Dong, J. W. Shao, C. Q. Chen, H. Li, R. X. Wang, Y. W. Chi, X. M. Lin and G. N. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- Q. Q. Li, S. Zhang, L. M. Dai and L. S. Li, J. Am. Chem. Soc., 2012, 134, 18932–18935 CrossRef CAS PubMed.
- J. Kwon, S. H. Lee, K. H. Park, D. H. Seo, J. Lee, B. S. Kong, K. Kang and S. Jeon, Small, 2011, 7, 864–868 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic diagram of the preparative strategy for GQDs, AFM images of GQD-A and GQD-B, Raman spectra of GQD-A and GQD-B, UV-Vis absorbances of GQD-A and GQD-B. Electrical properties of elastomer nanocomposites. See DOI: 10.1039/c6ra21651j |
| This journal is © The Royal Society of Chemistry 2016 |