
A series of organic–inorganic hybrid materials consisting of flexible organic amine modified polyoxomolybdates: synthesis, structures and properties†
Chunhua Gonga,
Xianghua Zenga,
Chengfeng Zhua,
Jiahui Shua,
Pingxiu Xiaoa,
Hao Xua,
Lichun Liu *a,
Junyong Zhanga,
Qingdao Zeng*b and
Jingli Xie*ac
*a,
Junyong Zhanga,
Qingdao Zeng*b and
Jingli Xie*ac
aCollege of Biological, Chemical Science and Engineering, Jiaxing University, Jiaxing 314001, P. R. China. E-mail: jlxie@mail.zjxu.edu.cn; lichun.liu@mail.zjxu.edu.cn
bCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, 11 Zhongguancun Beiyitiao, Beijing, 100190, P. R. China. E-mail: zengqd@nanoctr.cn
cState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, P. R. China
First published on 2nd November 2016
Abstract
A series of organic–inorganic hybrid complexes based on different types of polyoxomolybdates and transition metal complexes, namely, [Zn2(TPMA)2(H2P2Mo5O23)]·11H2O (1), [Zn2(TPMA)2(Mo8O26)] (2), [Co2(TPMA)2(Mo8O26)] (3), [Ni2(TPMA)2(Mo8O26)(H2O)2] (4), [Ni2(TPMA)2(2-PA)(H2O)](PMo12O40) (5) [Cu2(TPMA)2(Mo8O26)] (6), 2[Cu(TPMA)(CrMo6(OH)6O18)]·H[Cu2(TPMA)2(CrMo6(OH)6O18)]·4H2O (7) (TPMA = Tris[(2-pyridyl)methyl]amine, 2-PA = 2-picolinic acid), have been successfully synthesized under hydrothermal conditions. All complexes were characterized by single-crystal X-ray structural analysis, powder X-ray diffraction, IR spectroscopy and TG analysis. All the complexes showed polyoxomolybdate-based zero-dimensional (0D) structures, and could be further extended into three-dimensional (3D) supramolecular frameworks through hydrogen bonding interactions. In addition, the electrochemical properties of complexes 1–7 have been investigated. Interestingly, some complexes have efficient photocatalytic activities to degradate pararosaniline hydrochloride dye molecules.
Introduction
The development of multifunctional polyoxometalates represents one of the enduring challenges in the field of organic–inorganic hybrid materials.1 As discrete molybdenum oxide clusters with structural and compositional diversities, polyoxomolybdates include isopolyoxomolybdates and heteropolyoxomolybdates, such as [Mo6O19]2−,2 [Mo7O24]6−,3 [Mo8O26]4−,4 [P2Mo5O23]6−,5 [PMo12O40]3−,6 [CrMo6(OH)6O18]3− (ref. 7) and so on. These polyoxomolybdates have a number of terminal and bridging O atoms, and can be modified by different metal ions and organic ligands to form organic–inorganic hybrid materials.8 As such, the hybrid materials have the dual characteristics of polyoxomolybdates and organic molecules and consequently, they received considerable attention due to their multi-functional properties, such as magnetism,9 catalysis,10 molecular recognition,11 electrochemical activity.12 However, to date, it still remains a challenge to rationally synthesize organic–inorganic hybrid materials because of a variety of variable factors such as metal ions, organic ligands, solvent, pH and temperature. Therefore, the optimal selection of metal ions and organic ligands plays a key role for the design and synthesis of corresponding functional organic–inorganic hybrid materials.Even though there are numerous examples in the syntheses of diverse organic–inorganic hybrid materials by controlling the pH value,13 reaction temperature,14 reaction time,15 synthesis technologies16 and so on, it is anticipated that more specific crystalline materials could be achieved by systematically studying the experimental parameters and reaction mechanism of these synthetic processes could be elucidated to some extent. To explore intriguing variety of architecture of hybrid materials, chemists have embarked on the combination of different transition metal complexes with POMs cluster, and already got some novel structures.17 Obviously, this strategy could synthesize a variety of organic–inorganic hybrid materials incorporating different POM systems.
In the previous literature reports, multidentate flexible ligands Tris[(2-pyridyl)methyl]amine (TPMA) and their derivatives chelated with transition metal ions always shows good potential applications,18 such as magnetism, catalytic activity and drug delivery. However, the combination of these ligands and polyoxometalates haven't been reported. Thus, in this work, Tris[(2-pyridyl)methyl]amine (TPMA) was selected as the multidentate flexible ligand to assemble with different metal ions and various polyoxomolybdates, with the aim for constructing novel organic–inorganic hybrid materials (Scheme 1). Notably, a series of materials consisting of flexible organic amine (TPMA) modified polyoxomolybdates were obtained: [Zn2(TPMA)2(H2P2Mo5O23)]·11H2O (1), [Zn2(TPMA)2(Mo8O26)] (2), [Co2(TPMA)2(Mo8O26)] (3), [Ni2(TPMA)2(Mo8O26)(H2O)2] (4), [Ni2(TPMA)2(2-PA)(H2O)](PMo12O40) (5), [Cu2(TPMA)2(Mo8O26)] (6), 2[Cu(TPMA)(CrMo6(OH)6O18)]·H[Cu2(TPMA)2(CrMo6(OH)6O18)]·4H2O (7). All complexes 1–7 shown polyoxomolybdates-based zero-dimensional (0D) structures, and could be further extended into 3D supramolecular frameworks through hydrogen bonding interactions. In addition, the electrochemical properties of complexes 1–7 have been investigated. Moreover, some complexes have efficient photocatalytic activities to degradate pararosaniline hydrochloride dye molecule.
 | ||
| Scheme 1 The main ligand (TPMA), auxiliary ligand (2-PA) and extra auxiliary reagent (imidazole). | ||
Experimental
Materials and methods
All the chemicals were received as reagent grade and used without any further purification. The ligand TPMA and Na3[CrMo6(OH)6O18]·8H2O were synthesized according to literatures.19 The IR spectra were obtained on a Varian 640 FT/IR spectrometer with KBr pellet in 400–4000 cm−1 region. Powder X-ray diffraction (PXRD) was performed on a DX-2600 spectrometer. The thermal gravimetric analyses (TGA) were carried out in flowing N2 on a SDT 2960 differential thermal analyzer with a rate of 10 °C min−1. Cyclic voltammograms were obtained with a CHI 440A electrochemical workstation at room temperature. Platinum wire was used as a counter electrode and Ag/AgCl electrode was used as a reference electrode. Chemically bulk-modified carbon paste electrode (CPE) was used as the working electrode. The UV experiments were carried out on a Thermo EV 201CP.Syntheses of TPMA ligand and complexes 1–7
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2/MeOH). The final product was brown solid powder (5.51 g, 65.7% yield).
:1 CH2Cl2/MeOH). The final product was brown solid powder (5.51 g, 65.7% yield).
 | ||
| Scheme 2 Synthetic route for the TPMA ligand. | ||
Preparation of complexes 1–7 bulk-modified CPEs
The preparation method:20 the bulk-modified Carbon Paste Electrode (CPE) was fabricated by mixing 100 mg graphite powder and 10 mg target compound in an agate mortar for approximately 30 min to achieve a uniform mixture; then two drops methylsilicone oil was added and stirred with a copper rod. The homogenized mixture was packed into a 2 mm inner diameter glass tube and the tube surface was wrapped with weighing paper. The electrical contact was established with the copper wire through the back of the electrode. The bare CPE was prepared by similar process without target complexes.X-ray crystallography
Single crystal X-ray diffraction analysis data for complexes 1–7 were collected with an Oxford Diffraction Gemini R Ultra diffractometer with graphite-monochromated Mo-Kα (λ = 0.71073 Å) or Cu-Kα (λ = 1.54184 Å) at 296 K. All structures were solved by direct methods and refined on F2 by full-matrix least-squares methods using the SHELXTL package.21 In the structure of compound 1, hydrogen atoms attached to water molecules with partial site occupancy were not located. A summary of the crystal data and structure refinements of complexes 1–7 are provided in Table 1. Selected bond lengths and angles are listed in Table S1.† Crystallographic data for complexes 1–7 have been deposited in the Cambridge Crystallographic Data Center, their crystal structures can be obtained with CCDC reference numbers 1471018–1471020 (1–3), 1471022 (4), 1471021 (5), 1486195 (6), 1486075 (7), respectively.| Complexes | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = ∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]1/2. | ||||
| Empirical formula | C36H60Mo5N8O34P2Zn2 | C36H36Mo8N8O26Zn2 | C36H36Mo8N8O26Co2 | C36H40Mo8N8O28Ni2 |
| Formula weight | 1821.30 | 1894.99 | 1882.11 | 1917.67 |
| Temperature/K | 296(2) | 296(2) | 296(2) | 296(2) |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P21/c | Pbca | Pbca | P21/n |
| a/Å | 11.8566(5) | 16.6740(4) | 16.7082(3) | 12.2553(4) |
| b/Å | 19.7359(8) | 15.9270(3) | 15.9095(3) | 15.3666(4) |
| c/Å | 26.2178(10) | 19.1977(4) | 19.2987(3) | 15.4604(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 100.164(4) | 90 | 90 | 110.489(4) |
| γ/° | 90 | 90 | 90 | 90 |
| Volume/Å3 | 6038.7(4) | 5098.28(19) | 5129.96(17) | 2727.36(14) |
| Z | 4 | 4 | 4 | 2 |
| ρcalc/g cm−3 | 2.003 | 2.469 | 2.437 | 2.3349 |
| μ/mm−1 | 1.938 | 2.915 | 2.609 | 2.540 |
| F(000) | 3616 | 3648 | 3624 | 1830.6 |
| Radiation | MoKα | MoKα | MoKα | MoKα |
| Reflections collected | 24542 |
12810 |
15244 |
12597 |
| Independent reflections | 10612 |
4498 | 5988 | 4794 |
| Rint | 0.0465 | 0.0226 | 0.0215 | 0.0273 |
| GOOF | 1.127 | 1.055 | 1.057 | 1.056 |
| R1,a wR2b [I ≥ 2σ(I)] | 0.0731, 0.1708 | 0.0214, 0.0472 | 0.0237, 0.0508 | 0.0292, 0.0634 |
| R1, wR2 [all data] | 0.0942, 0.1789 | 0.0291, 0.0495 | 0.0340, 0.0547 | 0.0394, 0.0692 |
| Complexes | 5 | 6 | 7 |
|---|---|---|---|
| Empirical formula | C42H42Mo12N9O43PNi2 | C36H36Mo8N8O26Cu2 | C72H99Mo18N16O76Cr3Cu4 |
| Formula weight | 2660.48 | 1891.33 | 4541.75 |
| Temperature/K | 296(2) | 296(2) | 296(2) |
| Crystal system | Monoclinic | Orthorhombic | Triclinic |
| Space group | P21/c | Pbca | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 11.2559(4) | 16.8760(3) | 14.4947(6) |
| b/Å | 24.1685(11) | 15.9080(3) | 15.7549(4) |
| c/Å | 25.7463(9) | 18.9788(3) | 15.8183(6) |
| α/° | 90 | 90 | 99.314(3) |
| β/° | 100.287(4) | 90 | 116.519(4) |
| γ/° | 90 | 90 | 91.546(3) |
| Volume/Å3 | 6891.4(5) | 5095.10(15) | 3169.4(2) |
| Z | 4 | 4 | 1 |
| ρcalc/g cm−3 | 2.564 | 2.466 | 2.380 |
| μ/mm−1 | 18.982 | 2.81 | 2.718 |
| F(000) | 5104 | 3640 | 2195 |
| Radiation | CuKα | MoKα | MoKα |
| Reflections collected | 29399 |
15971 |
23137 |
| Independent reflections | 13356 |
5920 | 11152 |
| Rint | 0.0561 | 0.0276 | 0.0227 |
| GOOF | 1.059 | 1.051 | 1.021 |
| R1, wR2 [I ≥ 2σ(I)] | 0.0525, 0.1122 | 0.0260, 0.0521 | 0.0280, 0.0610 |
| R1, wR2 [all data] | 0.0935, 0.1315 | 0.0386, 0.0582 | 0.0392, 0.0651 |
Results and discussion
Structural description of complexes 1–7
In the structure of 1, there are two crystallographically independent Zn(II) ions (Fig. 1a). Zn1 ion is five-coordinated in a triangular bipyramid configuration by one O atom from P2Mo5 polyoxoanion (Mo–O) and four N atoms from TPMA ligand, in which three N atoms from pyridine generate an approximate triangle plane, and O atom and the fourth N atom serve as vertices. Zn2 ion is also five-coordinated in a distorted triangular bipyramid geometry by four N atoms from another TPMA ligand and one O atom from the same P2Mo5 polyoxoanion (P–O). The bond lengths ranges of Zn–O and Zn–N are 1.917(6)–1.977(5) Å and 2.066(8)–2.245(7) Å, respectively (Table S1†). The unit of compound 1 are connected by intermolecular hydrogen bonds [C(26)–H(26)⋯O(16), 3.234(11) Å] into a 1D supramolecular left-handed chain, and the adjacent chains are right-handed chains, therefore compound 1 is achiral complex (Fig. 1b and c). Then adjacent 1D chains are further linked into 2D supramolecular layer through hydrogen bonds [C(36)–H(36)⋯O(22), 3.205(12) Å] (Fig. 1d). 2D layers are extended into a 3D supramolecular framework through H-bonding interaction [C(30)–H(30)⋯O(15), 3.363(12) Å] (Fig. 1e).
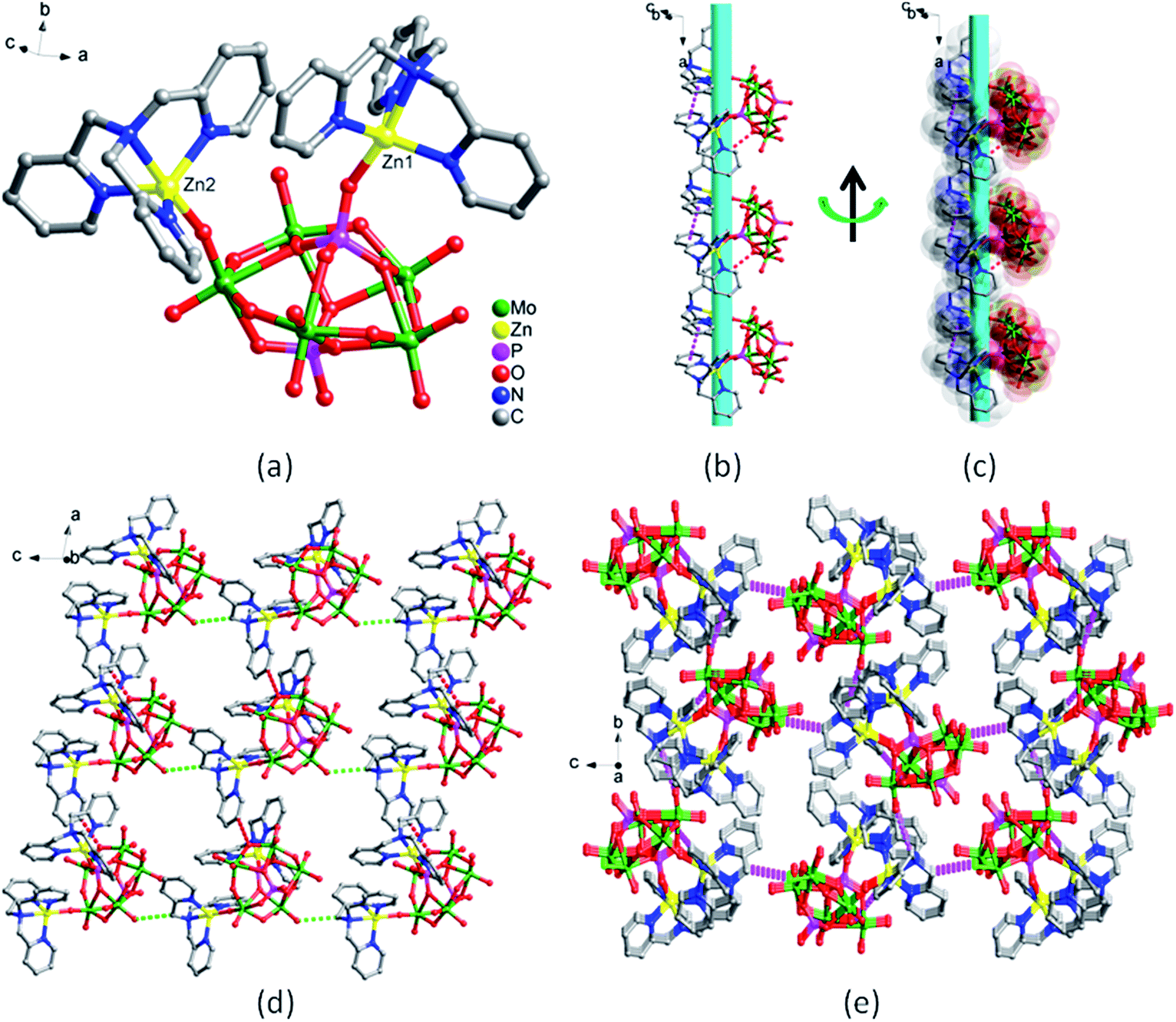 | ||
| Fig. 1 The structure of compound 1: (a) coordination environments for Zn ions and P2Mo5 polyoxoanion (the hydrogen atoms are omitted for clarity). (b) and (c) The ball-and-stick and space-filling view of left-handed 1D chain by hydrogen bonding interactions along black arrow direction, respectively. (d) 2D supramolecular layer and (e) 3D supramolecular framework formed by hydrogen bonding interactions. | ||
In compound 2, the coordination environments of the two Zn(II) ions are similar to each other (Fig. 2a). Zn ion is five-coordinated in a triangular bipyramid geometry by four N atoms from TPMA ligand and one O atom from Mo8 polyoxoanion (Mo–O). The bond lengths ranges of Zn–O and Zn–N are 1.998(2) Å and 2.046(3)–2.219(3) Å, respectively (Table S1†). Interestingly, it is worth mentioning that the Mo8 structure is transformed from the starting material CrMo6 and this kind of transformation between different POM structures have been reported in the literatures.23
 | ||
| Fig. 2 The structure of compound 2. (a) Coordination environments for Zn ions and Mo8 polyoxoanion (the hydrogen atoms are omitted for clarity). (b) 2D supramolecular layer by H bonds interactions. (c) 3D supramolecular framework by hydrogen bonding interactions. | ||
TPMA are coordinated with Zn(II) ions and then located on both sides of Mo8 to form a sandwich structure. The sandwich structures are connected by intermolecular hydrogen bonds [C(16)–H(16)⋯O(3), 3.182(4) Å] into a 2D supramolecular layer (Fig. 2b). 2D layers are extended into a 3D supramolecular framework through H-bonding interaction [C(6)–H(6B)⋯O(8), 3.359(4) Å] (Fig. 2c).
Complexes 2, 3 and 6 are isostructural and possess different metal ions. In compound 3, the bond lengths ranges of Co–O and Co–N are 1.9695(19) Å and 2.045(2) Å–2.208(2) Å, respectively (Table S1†). In compound 6, the bond lengths ranges of Cu–O and Cu–N are 1.930(2) Å and 2.037(3) Å–2.089(3) Å, respectively (Table S1†).
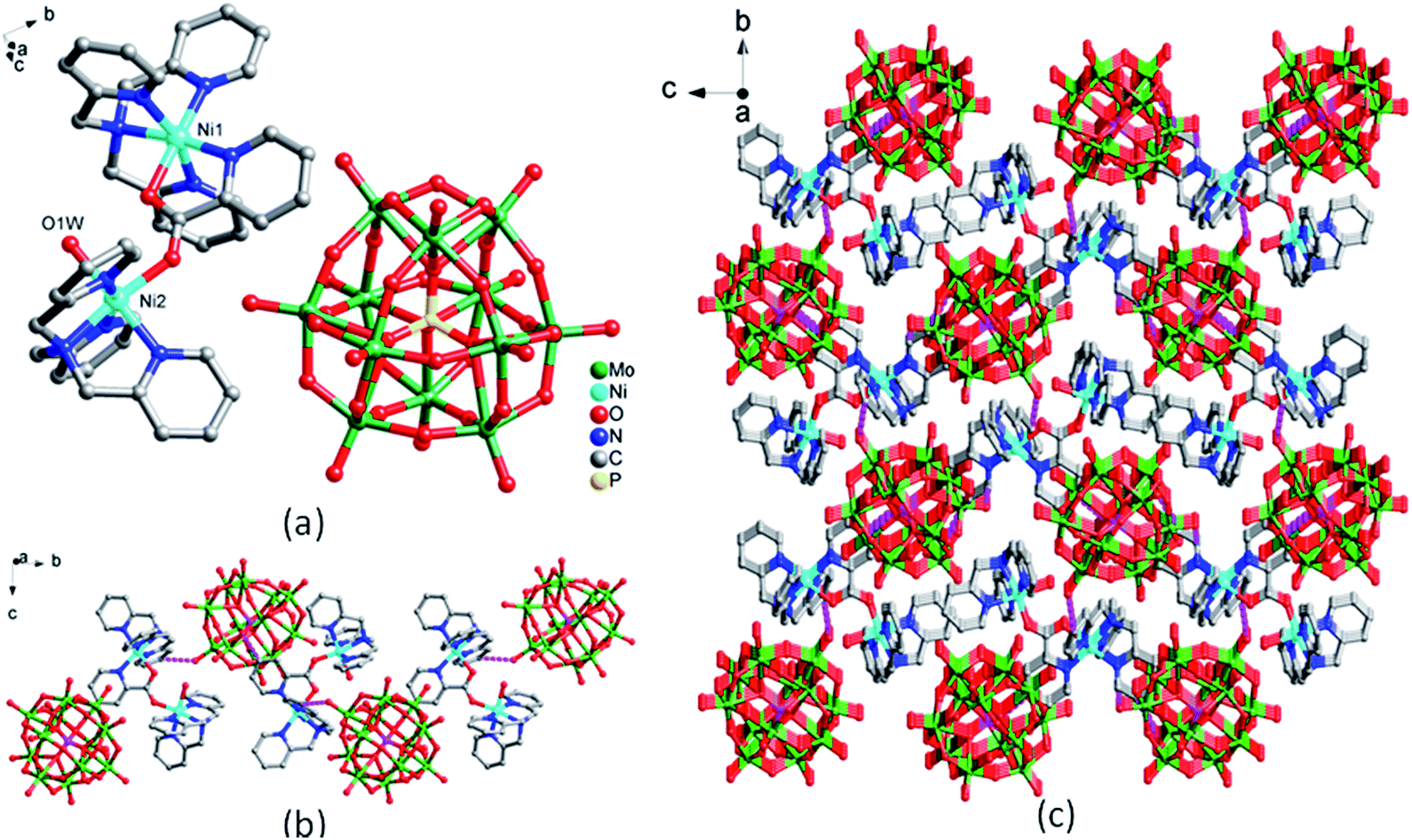
In compound 4, the coordination environments of the two Ni(II) ions are same (Fig. 3a). Ni ion is six-coordinated in a distorted octahedral geometry by four N atoms from TPMA ligand, one O atom from Mo8 polyoxoanion (Mo–O) and one O atom from water molecule. The bond lengths ranges of Cu–O and Cu–N are 1.943(4)–2.782(5) Å and 1.945(5)–1.993(5) Å, respectively (Table S1†). TPMA are coordinated with Ni(II) ions and then located on both sides of Mo8 to form a sandwich structure. The sandwich structures are connected by intermolecular hydrogen bonds [C(16)–H(16)⋯O(13), 3.102(4) Å] into a 2D supramolecular layer (Fig. 3b). 2D layers are extended into a 3D supramolecular framework through H-bonding interaction [C(2)–H(2)⋯O(12), 3.171(4) Å] (Fig. 3c).
 | ||
| Fig. 3 The structure of complex 4. (a) Coordination environments for Ni ions and Mo8 polyoxoanion (the hydrogen atoms are omitted for clarity). (b) 2D supramolecular layer by H bonds interactions. (c) 3D supramolecular framework by H bonds interactions. | ||
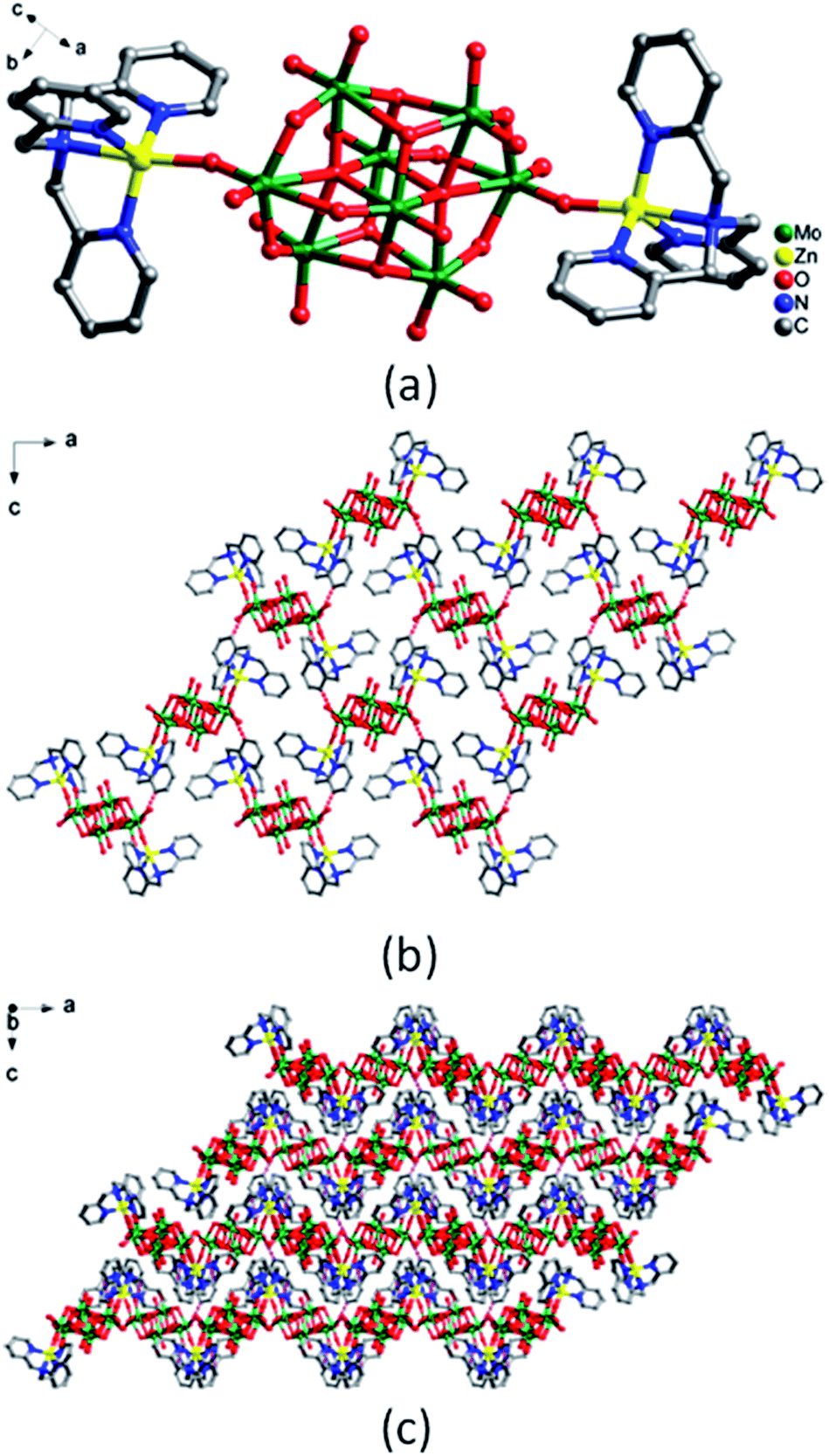
In complex 5, the coordination environments of the two Ni(II) ions are different (Fig. 4a). Ni1 ion is six-coordinated in a distorted octahedral geometry by four N atoms from TPMA ligand, one N atom and one O(O–H) atom from 2-PA. Ni2 ion is six-coordinated in a distorted octahedral geometry by four N atoms from another TPMA ligand, one O(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) atoms from the same 2-PA and one O atom from water molecule. The bond lengths ranges of Ni–O and Ni–N are 2.002(7)–2.100(7) Å and 2.046(8)–2.112(9) Å, respectively (Table S1†).
O) atoms from the same 2-PA and one O atom from water molecule. The bond lengths ranges of Ni–O and Ni–N are 2.002(7)–2.100(7) Å and 2.046(8)–2.112(9) Å, respectively (Table S1†).
 | ||
| Fig. 4 The structure of compound 5. (a) Coordination environments for Ni ions (the hydrogen atoms are omitted for clarity). (b) ‘Z’-type 1D chain by H bonds interactions. (c) 3D supramolecular framework by H bonds interactions. | ||
Two Ni(II) ions are bridged by the carboxyl of 2-PA to produce a dinuclear Ni(II) cluster. The dinuclear Ni(II) cluster are connected by intermolecular hydrogen bonds [C(39)–H(39)⋯O(39), 3.145(11) Å; C(15)–H(15)⋯O(31), 3.128(11) Å] into a 1D supramolecular ‘Z’-type chain (Fig. 4b), and the adjacent chains are extended into a 3D supramolecular framework through H-bonding interaction [C(7)–H(7)⋯O(18), 3.127(12) Å; C(8)–H(8)⋯O(5), 3.125(12) Å] (Fig. 4c).
. Intriguingly, there are two kinds of independent structures and two interstitial water molecules in 7. The first structure consists of one [CrMo6(OH)6O18]3− (abbreviated as CrMo6) polyoxoanion, one TPMA ligands and one copper ion. The second structure consists of one CrMo6 polyoxoanion, two TPMA ligands and two copper ions. Bond valence sum calculations22 show that all molybdenum atoms are in the +6 oxidation state, chrome atoms are in the +3 oxidation state, and copper atoms are in the +2 oxidation state.In the first structure of compound 7, Cu1 ion is five-coordinated in a trigonal bipyramid geometry by four N atoms from TPMA ligand and one O atom from CrMo6 polyoxoanion. In the second structure of 7, the coordination environments of the two Cu ions are same and symmetry related (Fig. 5a), Cu2 ion is also five-coordinated in a trigonal bipyramid geometry by four N atoms from TPMA ligand and one O atom from CrMo6 polyoxoanion. The bond lengths ranges of Cu–O and Cu–N are 1.914(4) Å–1.994(4) Å and 2.018(6) Å–2.079(7) Å, respectively (Table S1†).
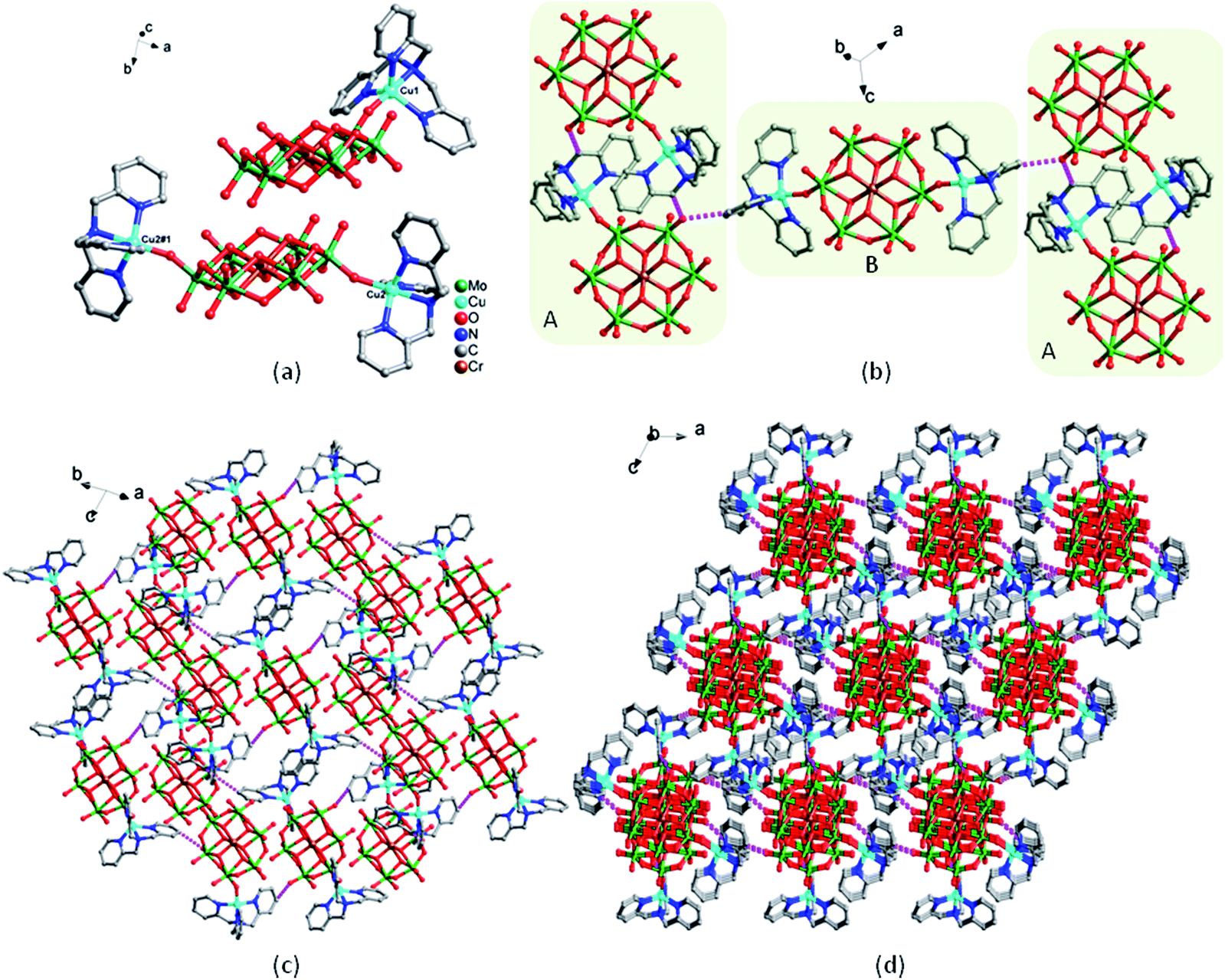 | ||
| Fig. 5 The structure of compound 7. (a) Coordination environments for Cu ions (the hydrogen atoms are omitted for clarity). (b) The 1D chain by H bonds interactions in the connection mode of ABAB. (c) 2D supramolecular layer and (d) 3D supramolecular framework connected by H bonds interactions. | ||
The two independent structures are linked by intermolecular hydrogen bonds [C(21)–H(21)⋯O(21), 3.024(11) Å] into a 1D supramolecular chain with the connection mode of ABAB (Fig. 5b), and the adjacent chains are connected by intermolecular hydrogen bonds [C(3)–H(3)⋯O(23), 3.074(11) Å] into a 2D supramolecular layer (Fig. 5c). Further, 2D layers are extended into a 3D supramolecular framework through H-bonding interaction [C(20)–H(20)⋯O(7), 3.156(12) Å] (Fig. 5d).
FT-IR spectra and powder X-ray diffraction
The IR spectra of complexes 1–7 are shown in Fig. S1.† For compound 1, the bands at 856, 793, 627 and 963 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot), νas(Mo–O–Mo) and νas(P–O). For compound 2, the bands at 1125, 826 and 750 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot) and νas(Mo–O–Mo). For compound 3, the bands at 985, 779 and 742 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot) and νas(Mo–O–Mo). For compound 4, the bands at 994, 829 and 728 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot) and νas(Mo–O–Mo). For compound 5, the bands at 936, 803, 728, 1090 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot), νas(Mo–O–Mo) and νas(P–O), the bands at 1619 cm−1 could be ascribed to the characteristic peaks of νas(CO) from the ancillary ligand of 2-PA. For compound 6, the bands at 963, 832 and 709 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot) and νas(Mo–O–Mo). For compound 7, the bands at 1016, 954 and 793 cm−1 could be ascribed to the characteristic peaks of νas(Mo–Ot) and νas(Mo–O–Mo). For 1–7, the bands in the region of 1700 to 1200 cm−1 could be ascribed to the characteristic peaks of the TPMA ligand. The bands in the region of 3750 to 3000 cm−1 could be ascribed to the characteristic peaks of the νas(–OH) from the water molecules.
To indicate the phase purities of complexes 1–7, PXRD experiments were carried out. As shown in Fig. S2,† diffraction peaks of both simulated and experimental patterns match well in the key positions, which indicate the phase purity of bulky samples. The differences in intensity may be aroused from the preferred orientation of the powder samples.24
Electrochemical properties
The electrochemical properties of 1–7 have been investigated in detail in 0.5 M Na2SO4 + 0.1 M H2SO4 aqueous solution (Fig. 6 and S4†). In the potential range from 0–500 mV, there are two pairs of reversible redox peaks (I–I′ and II–II′) for compound 1. The mean peak potentials E1/2 = (Ecp + Eap)/2 (scan rates: 200 mV s−1) are 273 and 83 mV, respectively. In the potential range from 0–500 mV, there are three pairs of reversible redox peaks (I–I′, II–II′, III–III′) for compound 2. The mean peak potentials E1/2 = (Ecp + Eap)/2 (scan rates: 200 mV s−1) are 394, 248 and 101 mV, respectively. While compound 3 shows two pairs of weak redox peaks. For compound 4, there are three pairs of reversible redox peaks (I–I′, II–II′, III–III′) in which the mean peak potentials E1/2 = (Ecp + Eap)/2 (scan rates: 200 mV s−1) are 372, 239 and 112 mV, respectively. In the potential range from −200 to 500 mV, there are three pairs of obvious reversible redox peaks (I–I′, II–II′, III–III′) for compound 5. The mean peak potentials E1/2 = (Ecp + Eap)/2 (scan rates: 200 mV s−1) are 344, 125 and −168 mV, respectively. In the potential range from 0–400 mV, there are two pairs of reversible redox peaks (I–I′ and II–II′) for 6, and their mean peak potentials E1/2 = (Ecp + Eap)/2 (scan rates: 200 mV s−1) are 222 and 110 mV, respectively. In the potential range from 100–600 mV, there is only one pair of weak redox peak (I–I′) for complex 7. The mean peak potentials E1/2 = (Ecp + Eap)/2 (scan rates: 200 mV s−1) is 221 mV. | ||
| Fig. 6 Cyclic voltammograms of 1–6 CPEs in 0.5 M Na2SO4 + 0.1 M H2SO4 aqueous solution at distinct scanning rates (from inside to out: 50, 100, 150, 200, 250, 300, 350, 400, 450, 500 mV s−1) respectively. | ||
As for 1–7, the redox peaks may arise from the redox process of Mo-based polyoxoanion clusters, but the pairs number of redox peaks are different which may be caused by the diverse coordination modes of those complexes.25 The cathodal peak potentials of complexes 1–7 move to the minus orientation, and the corresponding anodal peak potentials move toward the plus orientation with increasing scanning rates.
Photocatalytic activities
It is well known that a wide range of polyoxomolybdates possess photocatalytic activities in the degradations of organic dyes under UV irradiation.26 Thus, we explored the photocatalytic performances of complexes 1–7 for the photodegradation of pararosaniline hydrochloride with UV irradiation through a typical process: complexes 1–7 (20 mg) were added to the 50 mL aqueous solution of pararosaniline hydrochloride (6 mg L−1), respectively, and stirred for about 15 minutes to ensure the equilibrium of the resulting solution. Then the solution was exposed to UV irradiation from a 100 W Hg lamp (λ = 365 nm). The solution was kept for stirring during the irradiation. At every 30 min intervals, 5 mL of sample was taken out for UV analysis.As observed in Fig. 7, complexes 1, 4, 5, 6 and 7 display excellent degradation effect for pararosaniline hydrochloride. It is obvious that the absorption peaks of pararosaniline hydrochloride decreased significantly with illumination time. The calculation results show that the conversion of pararosaniline hydrochloride after irradiation in 3 hours is 93.25% for 1, 87.87% for 4, 76.48% for 5, 81.31% for 6, 90.16% for 7, respectively. Though complexes 2 and 3 have similar structures with complex 6, but their photocatalytic activities are far less than that of complex 6. As shown in Fig. S5† and 7f, their degradation efficiency for pararosaniline hydrochloride are only 31.71% and 32.17%, respectively, this result maybe caused by the difference of their center metal ions. By contrast, the order of photocatalytic activity is 1 > 7 > 4 > 6 > 5 (Fig. 7f). The photocatalytic behaviors of those five complexes indicate that they may be good photocatalysts for the photodegradation of organic dyes.
 | ||
| Fig. 7 Photocatalytic activities of complexes 1 (a), 4 (b), 5 (c), 6 (d), 7 (e) and the comparison for their degradation rates of complexes 1–7 (f), inside photo: before (left) and after (right) illumination. | ||
Conclusions
In this contribution, a series of organic–inorganic hybrid complexes based on different types of polyoxomolybdates and transition complexes have been successfully synthesized under hydrothermal conditions. Complexes 1–7 show polyoxomolybdates-based zero-dimensional (0D) structures, and can be further extended into three-dimensional (3D) supramolecular frameworks through hydrogen bonding interactions. Intriguingly, four kinds of polyoxometalates (P2Mo5, Mo8, PMo12, CrMo6) present in complexes 1–7 and this indicates the diversity of polyoxomolybdates. Additionally, the electrochemical properties of complexes 1–7 have been investigated. Moreover, some complexes have efficient photocatalytic activities to degradate pararosaniline hydrochloride dye molecule. It is believed that this work could provide useful information for the construction of multifunctional polyoxomolybdates-based organic–inorganic hybrid materials and various architectures of those materials could be observed consistently.Acknowledgements
Financial support from the National Natural Science Foundation of China (21371078, 21401077, 21472029), the National Key Basic Research Program of China (2013CB934200) is gratefully acknowledged.Notes and references
- (a) S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed; (b) J. M. Clemente-Juan, E. Coronado and A. Gaita-Arino, Chem. Soc. Rev., 2012, 41, 7464 RSC; (c) S. M. Lauinger, J. M. Sumliner, Q. S. Yin, Z. H. Xu, G. J. Liang, E. N. Glass, T. Q. Lian and C. L. Hill, Chem. Mater., 2015, 27, 5886 CrossRef CAS; (d) J. W. Sun, P. F. Yan, G. H. An, J. Q. Sha, C. Wang and G. M. Li, Dalton Trans., 2016, 45, 1657 RSC; (e) M. Singh and A. Ramanan, Cryst. Growth Des., 2011, 11, 3381 CrossRef CAS; (f) E. L. Zhou, C. Qin, P. Huang, X. L. Wang, W. C. Chen, K. Z. Shao and Z. M. Su, Chem.–Eur. J., 2015, 21, 1 CrossRef.
- (a) M. Che, M. Fournier and J. P. Launay, J. Chem. Phys., 1979, 71, 1954 CrossRef CAS; (b) M. X. Li, H. L. Chen, J. P. Geng, X. He, M. Shao, S. R. Zhu and Z. X. Wang, CrystEngComm, 2011, 13, 1687 RSC.
- (a) P. Hili, P. A. Lorenzo-Luis, A. Mederos, J. M. Arrieta, G. Germain, A. Castiñeiras and R. Carballo, Inorg. Chim. Acta, 1999, 295, 106 CrossRef; (b) Y. Z. Fu, J. Coord. Chem., 2010, 63, 1856 CrossRef CAS.
- (a) M. Filowitz, R. K. C. Ho, W. G. Klemperer and W. Shum, Inorg. Chem., 1979, 18, 93 CrossRef CAS; (b) Q. Li, Y. G. Wei, J. Hao, Y. L. Zhu and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 5810 CrossRef CAS PubMed; (c) C. Z. Lu, C. D. Wu, H. H. Zhuang and J. S. Huang, Chem. Mater., 2002, 14, 2649 CrossRef CAS.
- (a) W. Kwak, M. T. Pope and T. F. Scully, J. Am. Chem. Soc., 1975, 97, 5735 CrossRef CAS; (b) J. Y. Niu, J. C. Ma, J. W. Zhao, P. T. Ma and J. P. Wang, Inorg. Chem. Commun., 2011, 14, 474 CrossRef CAS.
- (a) H. J. Wu, Biol. Chem., 1920, 43, 189 CAS; (b) X. Wang, J. Peng, K. Alimaje and Z. Y. Shi, CrystEngComm, 2012, 14, 8509 RSC.
- (a) V. Lee and Y. Sasaki, Chem. Lett., 1984, 1297 CrossRef; (b) M. Singh, S. E. Lofland, K. V. Ramanujachary and A. Ramanan, Cryst. Growth Des., 2010, 10, 5105 CrossRef CAS.
- (a) C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS PubMed; (b) D. B. Dang, B. An, Y. Bai and J. Y. Niu, Dalton Trans., 2012, 41, 13856 RSC; (c) H. Y. Liu, J. Yang, Y. Y. Liu and J. F. Ma, Dalton Trans., 2012, 41, 9922 RSC; (d) Y. W. Liu, S. M. Liu, D. F. He, N. Li, Y. J. Ji, Z. P. Zheng, F. Luo, S. X. Liu, Z. Shi and C. W. Hu, J. Am. Chem. Soc., 2015, 137, 12697 CrossRef CAS PubMed; (e) S. Zhou, B. Liu, T. Shi, X. M. Li and Y. G. Chen, Inorg. Chem., 2015, 54, 7165 CrossRef CAS PubMed; (f) A. B. Lysenko, G. A. Senchyk, L. V. Lukashuk, K. V. Domasevitch, M. Handke, J. Lincke, H. Krautscheid, E. B. Rusanov, K. W. Kramer, S. Decurtins and S. X. Liu, Inorg. Chem., 2016, 55, 239 CrossRef CAS PubMed; (g) L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, A. Sampieri, D. W. Lewis, A. Gómez, B. Nohra, P. Mialane, J. Marrot, F. Sécheresse, C. Mellot-Draznieks, R. N. Biboum, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2009, 131, 16078 CrossRef PubMed; (h) F. Y. Li and L. Xu, Dalton Trans., 2011, 40, 4024 RSC; (i) X. X. Li, Y. X. Wang, R. H. Wang, C. Y. Cui, C. B. Tian and G. Y. Yang, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef PubMed.
- (a) Y. M. Xie, R. M. Yu, X. Y. Wu, F. Wang, J. Zhang and C. Z. Lu, Cryst. Growth Des., 2011, 11, 4739 CrossRef CAS; (b) X. Y. Wu, P. Dong, R. M. Yu, Q. K. Zhang, X. F. Kuang, S. C. Chen, Q. P. Lin and C. Z. Lu, CrystEngComm, 2011, 13, 3686 RSC; (c) A. B. Khélifa, M. S. Belkhiria, G. Huang, S. Freslon, O. Guilloub and K. Bernot, Dalton Trans., 2015, 44, 16458 RSC.
- (a) H. Q. Tan, W. L. Chen, D. Liu, X. J. Feng, Y. G. Li, A. X. Yan and E. B. Wang, Dalton Trans., 2011, 40, 8414 RSC; (b) Y. Q. Chen, G. R. Li, Y. K. Qu, Y. H. Zhang, K. H. He, Q. Gao and X. H. Bu, Cryst. Growth Des., 2013, 13, 901 CrossRef CAS; (c) G. A. Senchyk, A. B. Lysenko, A. A. Babaryk, E. B. Rusanov, H. Krautscheid, P. Neves, A. A. Valente, I. S. Goncalves, K. W. Kramer, S. X. Liu, S. Decurtins and K. V. Domasevitch, Inorg. Chem., 2014, 53, 10112 CrossRef CAS PubMed; (d) A. B. Lysenko, G. A. Senchyk, K. V. Domasevitch, J. Hauser, D. Fuhrmann, M. Kobalz, H. Krautscheid, P. Neves, A. A. Valente and I. S. Goncalves, Inorg. Chem., 2015, 54, 8327 CrossRef CAS PubMed.
- (a) X. L. Wang, N. Li, A. X. Tian, J. Ying, T. J. Li, X. L. Lin, J. Luan and Y. Yang, Inorg. Chem., 2014, 53, 7118 CrossRef CAS PubMed; (b) A. X. Tian, Y. L. Ning, Y. Yang, X. Hou, J. Ying, G. C. Liu, J. W. Zhang and X. L. Wang, Dalton Trans., 2015, 44, 16486 RSC.
- (a) A. Dolbecq, P. Mialane, F. Sécheresse, B. Keita and L. Nadjo, Chem. Commun., 2012, 48, 8299 RSC; (b) S. B. Li, L. Zhang, H. Y. Ma, H. J. Pang and C. Y. Zhao, New J. Chem., 2015, 39, 3528 RSC.
- (a) Y. N. Chi, F. Y. Cui, A. R. Jia, X. Y. Ma and C. W. Hu, CrystEngComm, 2012, 14, 3183 RSC; (b) S. B. Li, H. Y. Ma, H. J. Pang and L. Zhang, Cryst. Growth Des., 2014, 14, 4450 CrossRef CAS; (c) S. Roy, S. Sarkar, J. Pan, U. V. Waghmare, R. Dhanya, C. Narayana and S. C. Peter, Inorg. Chem., 2016, 55, 3364 CrossRef CAS PubMed.
- (a) J. Y. Niu, J. A. Hua, X. Ma and J. P. Wang, CrystEngComm, 2012, 14, 4060 RSC; (b) M. L. Han, X. H. Chang, X. Feng, L. F. Ma and L. Y. Wang, CrystEngComm, 2014, 16, 1687 RSC; (c) H. F. Zhou, T. He, K. F. Yue, Y. L. Liu, C. S. Zhou, N. Yan and Y. Y. Wang, Cryst. Growth Des., 2016, 16, 3961 CrossRef CAS.
- (a) J. W. Zhang, X. M. Kan, B. Q. Liu, G. C. Liu, A. X. Tian and X. L. Wang, Chem.–Eur. J., 2015, 21, 16219 CrossRef CAS PubMed; (b) P. K. Dutta, S. Panda, G. R. Krishna, C. M. Reddy and S. S. Zade, Dalton Trans., 2013, 42, 476 RSC.
- (a) A. B. Lago, R. Carballo, S. Rodríguez-Hermida and E. M. Vázquez-López, Cryst. Growth Des., 2014, 14, 3096 CrossRef CAS; (b) G. D. Zou, Z. Z. He, C. B. Tian, L. J. Zhou, M. L. Feng, X. D. Zhang and X. Y. Huang, Cryst. Growth Des., 2014, 14, 4430 CrossRef CAS; (c) J. Y. Zhang, C. H. Gong, X. H. Zeng and J. L. Xie, Coord. Chem. Rev., 2016, 324, 39 CrossRef CAS; (d) J. Y. Zhang, S. Q. Chang, B. H. R. Suryanto, C. H. Gong, X. H. Zeng, C. Zhao, Q. D. Zeng and J. L. Xie, Inorg. Chem., 2016, 55, 5585 CrossRef CAS PubMed.
- (a) S. D. Han, J. P. Zhao, Y. Q. Chen, S. J. Liu, X. H. Miao, T. L. Hu and X. H. Bu, Cryst. Growth Des., 2014, 14, 2 CrossRef CAS; (b) J. H. Qin, Y. Y. Jia, H. J. Li, B. Zhao, D. Q. Wu, S. Q. Zang, H. W. Hou and Y. T. Fan, Inorg. Chem., 2014, 53, 685 CrossRef CAS PubMed; (c) P. P. Zhu, L. J. Sun, N. Sheng, J. Q. Sha, G. D. Liu, L. Yu, H. B. Qiu and S. X. Li, Cryst. Growth Des., 2016, 16, 3215 CrossRef CAS; (d) X. L. Wang, R. Zhang, X. Wang, H. Y. Lin and G. C. Liu, Inorg. Chem., 2016, 55, 6384 CrossRef CAS PubMed; (e) W. L. Sun, J. Yu, H. J. Pang, H. Y. Ma and C. Y. Zhao, New J. Chem., 2016, 40, 2944 RSC; (f) C. J. Wang, T. T. Wang, Q. Lan, S. Yao, H. L. Wu, Y. Y. Zhou, Z. M. Zhang and E. B. Wang, RSC Adv., 2016, 6, 15513 RSC.
- (a) O. Hietsoi, P. W. Dunk, H. D. Stout, A. Arroyave, K. Kovnir, R. E. Irons, N. Kassenova, R. Erkasov, C. Achim and M. Shatruk, Inorg. Chem., 2014, 53, 13070 CrossRef CAS PubMed; (b) A. Kaur, T. G. Ribelli, K. Schröder, K. Matyjaszewski and T. Pintauer, Inorg. Chem., 2015, 54, 1474 CrossRef CAS PubMed; (c) W. T. Eckenhoff, A. B. Biernesser and T. Pintauer, Inorg. Chem., 2012, 51, 11917 CrossRef CAS PubMed; (d) K. Schröder, R. T. Mathers, J. Buback, D. Konkolewicz, A. J. D. Magenau and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1037 CrossRef; (e) I. C. A. de Souza, L. V. Faro, C. B. Pinheiro, D. T. G. Gonzaga, F. de C. da Silva, V. F. Ferreira, F. da S. Miranda, M. Scarpellinic and M. Lanznaster, Dalton Trans., 2016, 45, 13671 RSC; (f) S. M. W. Rahaman, K. Matyjaszewskib and R. Poli, Polym. Chem., 2016, 7, 1079 RSC.
- (a) A. Beni, A. Dei, S. Laschi, M. Rizzitano and L. Sorace, Chem.–Eur. J., 2008, 14, 1804 CrossRef CAS PubMed; (b) A. Perloff, Inorg. Chem., 1970, 9, 2228 CrossRef CAS.
- X. L. Wang, Z. H. Kang, E. B. Wang and C. W. Hu, Mater. Lett., 2002, 56, 393 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- (a) H. X. Yang, S. Y. Gao, J. Lü, B. Xu, J. X. Lin and R. Cao, Inorg. Chem., 2010, 49, 736 CrossRef CAS PubMed; (b) D. B. Dang, B. An, Y. Bai and J. Y. Niu, Dalton Trans., 2012, 41, 13856 RSC; (c) X. L. Wang, X. J. Liu, A. X. Tian, J. Ying, H. Y. Lin, G. C. Liu and Q. Gao, Dalton Trans., 2012, 41, 9587 RSC; (d) X. L. Wang, C. H. Gong, J. W. Zhang, G. C. Liu, X. M. Kan and N. Xu, CrystEngComm, 2015, 17, 4179 RSC.
- H. Y. Zhao, J. W. Zhao, B. F. Yang, H. He and G. Y. Yang, Cryst. Growth Des., 2013, 13, 5169 CAS.
- (a) H. J. Jin, B. B. Zhou, Y. Yu, Z. F. Zhao and Z. H. Su, CrystEngComm, 2011, 13, 585 RSC; (b) Z. C. Yue, H. J. Du, Y. Y. Niu and G. X. Jin, CrystEngComm, 2013, 15, 9844 RSC.
- (a) H. Y. Liu, L. Bo, J. Yang, Y. Y. Liu, J. F. Ma and H. Wu, Dalton Trans., 2011, 40, 9782 RSC; (b) X. P. Sun, Z. J. Liang, P. T. Ma, R. Ban, M. S. Jiang, D. D. Zhang, J. P. Wang and J. Y. Niu, Dalton Trans., 2015, 44, 17544 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1471018–1471020 (1–3), 1471022 (4), 1471021 (5), 1486195 (6) and 1486075 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra21603j |
| This journal is © The Royal Society of Chemistry 2016 |