
Mechano-chemical activation of the (3LiBH4 + TiF3) system, its dehydrogenation behavior and the effects of ultrafine filamentary Ni and graphene additives†
Amirreza Shirani Bidabadia,
Robert A. Varin*a,
Marek Polanskib and
Leszek Stobinskic
aDepartment of Mechanical and Mechatronics Engineering, Waterloo Institute for Nanotechnology, University of Waterloo, 200 University Ave. W., Waterloo, Ontario, Canada N2L 3G1. E-mail: robert.varin@uwaterloo.ca
bFaculty of Advanced Technology and Chemistry, Military University of Technology, 2 Kaliskiego Str., 00-908 Warsaw, Poland
cFaculty of Materials Science and Engineering, Graphene Laboratory, Warsaw University of Technology, Warynskiego 1, 00-645 Warsaw, Poland
First published on 21st September 2016
Abstract
The influence of milling energy input, QTR (kJ g−1), during ball milling and additives such as ultrafine filamentary Ni and graphene (reduced graphene oxide), on the occurrence of the solid-state mechano-chemical reaction and resulting microstructure, were investigated for the (3LiBH4 + TiF3) system. The new phases LiF and Ti are observed after injecting the energy input QTR = 72.8 kJ g−1 (1 h ball milling). A mechanical dehydrogenation phenomenon occurs during mechano-chemical reaction. The ultrafine filamentary Ni additive does not measurably accelerate the rate of mechanical dehydrogenation while the rate of mechanical dehydrogenation with graphene is initially slow and then dramatically increases up to 5 h ball milling (QTR = 364 kJ g−1). Thermal desorption of ball milled samples occurs at a very low temperature of 60 °C. The addition of 5 wt% filamentary Ni mildly reduces the apparent average activation energy for desorption. The highest average apparent activation energy of 95.2 ± 1.9 kJ mol−1 is exhibited by a sample with 5 wt% graphene milled for 1 h which dramatically decreases after 5 h ball milling. The X-ray diffraction intensity of the LiF and Ti peaks greatly increases after thermal dehydrogenation. The principal gas released during thermal dehydrogenation is hydrogen although the 1 h ball milled (QTR = 72.8 kJ g−1) sample shows a very small quantity of diborane gas, B2H6, which ceased to be released after 5 h ball milling. It clearly shows that the release of B2H6 during thermal dehydrogenation depends on the quantity of milling (mechanical) energy injected into the powder mixture. Differential scanning calorimetry measurements show exothermic peaks for all samples regardless of the milling energy input. The ball milled samples release H2 during long term storage at room temperature.
1. Introduction
In the past two decades a general consensus emerged in the scientific community that hydrogen (H2) could become a principal, future potential clean energy carrier which would eventually lead to the implementation of the world-wide hydrogen economy.1–3 That decisive shift from a fossil fuel based energy supply would be allowed by applying hydrogen (H2) in a great variety of industrial and commercial applications. In the transitional stage of transformation from fossil fuel energy to a hydrogen energy supply, a wide usage of fuel cells (FC), where hydrogen gas (H2) in contact with oxygen (O2) is converted into an electrical energy, must become a widespread reality. Furthermore, finding effective hydrogen storage systems for supplying FCs is another most important issue. There are three possible H2 storage/generation methods: gaseous H2, liquid H2 and solid hydrides. The most attractive storage method is the third one in solid hydrides but it is also the most challenging, particularly, for mass transportation using cars (automotive).4 A proton exchange membrane fuel cell (PEM FC) stack (sometimes also named a Polymer Electrolyte Fuel Cell (PEFC)), which has been a primary candidate for an automotive power plant, imposes severe constraints on a solid state H2 hydride storage system such as a low dehydrogenation temperature range of 70–80 °C at 1.1–1.8 bar H2 pressure.5 Furthermore, the Department of Energy US (D.O.E) 2017–2020 targets for the driving distance of, at least, 300 miles (480 km) require a storage system with the gravimetric system capacity of at least 5.5 wt% H2 which translates to the storing hydride material capacity of roughly 11 wt% H2.5,6 In addition, the convenience of operating a Fuel Cell Vehicle (FCV) calls for reversible and quick “on board” refueling at a hydrogen station.Unfortunately, no solid hydride system that could meet all those requirements has been discovered in the past two decades of intensive research. Since the automotive solid state H2 storage is still very futuristic, the first commercial Toyota and Hyundai Fuel Cell Vehicles (FCV), coming to market in 2016–2017 store H2 in a more conventional, gaseous form under a high pressure of 70 MPa.7,8
However, there is a number of other potential market applications for simple H2 generation systems rather than “on board” reversible storage systems, where some of them could be even recharged “off board”, supplying H2 at the ambient and slightly elevated temperatures, in the commercial, non-automotive sectors of the economy. For example, they could be utilized for supplying fuel cells in such applications as stationary auxiliary power systems, portable electronic devices, off-road vehicles, lawn mowers, auxiliary devices in air transportation, coastal and international shipping, bulk hydrogen storage and many others.
There is a number of complex hydrides exhibiting high, theoretical H2 capacity, that are not reversible “on board” but some of them could be rechargeable “off board” which would serve well as simple one-way H2 generators. One of them is titanium borohydride, Ti(BH4)3. Its excellent theoretical gravimetric H2 capacity is 13.1 wt% (molar mass 92.39 g mol−1). The first time synthesis of Ti(BH4)3 was reported by Hoekstra and Katz9 using such reactants as lithium borohydride (LiBH4) and titanium tetrachloride (TiCl4) at a low temperature of −45 °C under vacuum. The obtained product was a green solid with a calculated empirical formula of Ti1.00B2.96H12.00 (Ti(BH4)3). Interestingly, the authors reported a release of diborane, B2H6, gas during the synthesis reaction. The successful synthesis of Ti(BH4)3 was of a great importance because, for the first time, it showed that Ti(BH4)3 could exist as a solid material at low temperatures and quickly decompose close to room temperature. Jensen et al.10 synthesized the diadducts Ti(BH4)3(PMe3)2 and Ti(BH4)3(PEt3)2 by the reaction of trialkylphosphines with thermally unstable Ti(BH4)3·Et2O, which was prepared in diethyl ether (Et2O) from the LiBH4 and TiCl4 reactants. These complexes were stable at room temperature for a few days. The authors managed to obtained the XRD data for the Ti(BH4)3(PMe3)2 adduct: monoclinic, space group Pnma, a = 10.757(1), b = 11.145(2) and c = 14.270(3) Å, and density of 0.950 g cm−3. From the obtained density and molar mass of 92.39 g mol−1 one can roughly estimate the volumetric H2 capacity of Ti(BH4)3 as being equal to about 124 kg H2 per m3 which is a very high volumetric capacity, indeed. More recently, Soloveichik reported11 the synthesis of Ti(BH4)3 by the reaction of LiBH4 with TiCl4 or TiCl3 and isolation by low temperature vacuum sublimation. He also mentioned that Ti fluoride salts did not react which was erroneous as will be shown later. The synthesized Ti(BH4)3 was a white volatile solid. Electron diffraction in the gas phase showed a monomer molecule with tridentate BH4− groups. The hydride was thermally unstable and decomposed to TiB2, H2, and B2H6 at 20 °C.11
A few authors added a small amount of halides such as TiCl3 and TiF3 to LiBH4 (ref. 12–15) with the aim of destabilizing LiBH4 and reduce its high dehydrogenation temperature.16 It has been observed that TiF3 reduced the onset of dehydrogenation temperature of the investigated systems. The literature on this particular hydride system is very scare. Only two papers have been published17,18 on the LiBH4–TiF3 system, a paper14 on the LiBH4–TiF3–SiO2 and one19 on the LiBH4–TiF3–Fe2O3 system which will be discussed thoroughly later on together with the obtained results of the present work.
Apparently, data and their interpretation reported in the available literature on the (3LiBH4 + TiF3) system are at least unclear and on occasion quite contradictory. Furthermore, no behavior of the (3LiBH4 + TiF3) mixture during ball milling, e.g. the phenomenon of mechanical dehydrogenation,3,5,20,21 has ever been investigated as a function of the milling energy input (kJ g−1). In view of the number of discrepancies in the published data and the overall scarcity of published papers on the (3LiBH4 + TiF3) system, the major scientific objective of this work is to investigate and understand in more detail the H2 generation behavior from the LiBH4 and TiF3 system in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio during ball milling with varying milling energy inputs and during subsequent thermolysis. In addition, we are investigating the effect of potential catalytic additives such as ultrafine filamentary nickel (Ni) which, in its submicrometric and nanometric form, has been found to be a very effective catalyst for metal and complex hydride systems2,3,5 and graphene which was recently investigated as an additive for improving sorption behavior of LiBH4.22 The (3LiBH4 + TiF3) system is quite attractive as a potential efficient hydrogen generator at low temperatures and deserves a full understanding.
:1 stoichiometric ratio during ball milling with varying milling energy inputs and during subsequent thermolysis. In addition, we are investigating the effect of potential catalytic additives such as ultrafine filamentary nickel (Ni) which, in its submicrometric and nanometric form, has been found to be a very effective catalyst for metal and complex hydride systems2,3,5 and graphene which was recently investigated as an additive for improving sorption behavior of LiBH4.22 The (3LiBH4 + TiF3) system is quite attractive as a potential efficient hydrogen generator at low temperatures and deserves a full understanding.
2. Experimental
In total, three samples were synthesized from the as-received powders. The first sample was plain (no additive) composed of the commercial lithium borohydride (LiBH4) (95% purity) and titanium fluoride (TiF3) (98% purity) powders which were purchased from Alfa Aesar (Canada) and mixed in the molar ratio of 3 to 1. The second and third samples with the same 3:1 molar ratio were made with the addition of 5 wt% ultrafine filamentary carbonyl nickel (Ni) powder supplied by the Cnem Corp. (Canada) and 5 wt% of few layer reduced graphene oxide platelets (flakes) (FL-RGO), referred to as “graphene” in the text, obtained from Nanomaterials (http://www.nanomaterials.pl). The FL-RGO product consists of 2–3 stacked nanostructure graphene layers and contains ∼9.6 wt% of oxygen and ∼1 wt% H2 (the exact chemical elemental analysis can be found at http://www.nanomaterials.pl).
In order to avoid reactions between the powder samples and moisture or oxygen from air, all processes during preparation of the samples were handled in a glove box containing a moisture-absorbing Drierite granulated compound. Before handling, the glove box was purged a few times with high purity argon gas (99.999% purity).
Mechano-chemical activation synthesis (MCAS) of powder mixtures was carried out for 1 and 5 hours in an ultra-high purity hydrogen gas atmosphere (purity 99.999%: O2 < 2 ppm; H2O < 3 ppm; CO2 < 1 ppm; N2 < 6 ppm; CO < 1 ppm; THC < 1 ppm) at ∼300 kPa pressure in a magneto-mill Uni-Ball-Mill 5 manufactured by A.O.C. Scientific Engineering Pty Ltd., Australia.2,23,24 Two magnets positioned at 6 and 8 o'clock to create a strong impact mode (IMP68), at a distance from the milling vial of ∼10 and ∼2 mm, respectively, were used in the milling process. The ball-to-powder weight ratio (R) and a number of hardened steel balls vial (mass 65 g and 25 mm in diameter each) were 132 and 4 respectively. Milling occurred at a rotational speed 200 rpm under continuous cooling by an air fan. The possible release of hydrogen during ball milling was continuously monitored by the pressure increase in the milling vial measured by a pressure gage (the accuracy ± 0.1 wt% H2).
The Sieverts-type apparatus custom-built by A.O.C. Scientific Engineering Pty Ltd., Australia2,25 was used for evaluating the hydrogen thermal desorption. This apparatus, built entirely of 316 austenitic stainless steel, allows loading a powder sample into a stainless steel reactor in a glove box under high purity argon and its subsequent transfer to the main unit in a sealed reactor without any exposure to the environment. Samples with a nearly constant masses of 30–40 mg were used in a desorption test. Before starting the desorption test, the inner tubing of the apparatus was evacuated and purged four times with ultra-high purity hydrogen. The furnace of the apparatus was heated separately to the desired test temperature and subsequently inserted onto a tightly sealed powder sample reactor inside which an atmospheric pressure of 1 bar H2 was kept. The powder sample in the reactor usually reaches the furnace temperature within ∼400 s in the temperature range of 100–200 °C which is negligible compared to desorption completion time. Hence, the test can be considered as isothermal. Desorption curves were corrected for the hydrogen gas expansion due to increase in temperature. The amount of desorbed hydrogen was calculated from the ideal gas law as described in detail in ref. 2 and expressed in wt% with respect to the total weight of powder sample. The calibrated accuracy of desorbed hydrogen capacity is about ±0.1 wt% H2 and that of temperature reading and stabilization ±0.1 °C.
The apparent activation energy for volumetric hydrogen desorption was estimated using the registered dehydrogenation curves by applying a simple Arrhenius equation2 following Sandrock et al.:26
| k = koe−EA/RT | (1) |
k vs. 1000/RT.
A Bruker D8 X-ray diffractometer using a monochromated CuKα1 radiation (λ = 0.15406 nm) with an accelerating voltage of 40 kV and a current of 30 mA was used for investigating phase transformation and the crystalline properties of powders. A custom made brass holder with Cu/glass plates and Kapton window transmittable to X-rays in the upper part of that was used to hold the sample in the X-ray machine.
The Williamson–Hall method27 in the following form was used to evaluate the crystallite size of LiF and the lattice strain:
|
βcosθ = 0.9λ/D + 2Aεsinθ
| (2) |
cosθ vs. 2sinθ which were calculated for several peaks. The slope of βcosθ vs. 2sinθ provided the average lattice strain while the average crystallite size was estimated from the intersection of this line at sinθ = 0.
Morphological characteristics of powders before and after milling and subsequent thermal desorption were studied by the LEO 1550 high resolution, field emission scanning electron microscope (FESEM) employing a secondary electron mode (SE) with the accelerating voltage of 10 kV. In a glove box filled with high purity argon, samples were dispersed on a sticky carbon tape and pictures were taken under secondary electron mode (SE).
The Fourier transform infrared spectroscopy (FT-IR) measurements were performed with a Nicolet 6700 apparatus at room temperature in the wavenumber range 600–3000 cm−1 to examine the features of chemical bonding states of samples. The FT-IR apparatus was put in a glove bag and purged continuously with high purity (5 N) nitrogen. The glove bag was opened and the glass vial containing a powder sample was inserted into a glove bag. After closing the glove bag the sample was dispersed onto the sample holder and inserted in the machine under nitrogen atmosphere. Subsequently, further measurements were carried out under the atmosphere of high purity (5 N) nitrogen gas. The measurements were carried out in DRIFTS mode. The raw data, with automatic background subtraction, were plotted. The resolution was 4 cm−1 for all data and the units on the plots are Kubelka–Munk units.
The differential scanning calorimetry (DSC) analysis was conducted simultaneously with the thermogravimetric analysis (TGA) on a Setaram Sensys Evo 3d analyzer (France). The analyzer was coupled with a quadrupole mass spectrometer Hiden Analytical (United Kingdom). Each powdered sample (∼10–30 mg) was loaded into an alumina crucible of 100 μl volume and covered with alumina powder almost to the top of the crucible to prevent the oxidation and hydrolysis during the quick transfer to the analyzer and also to avoid a volatile foaming and flowing out of the crucible if the powder sample melted. After loading to the analyzer, each sample was flushed with high purity helium gas (<10 ppm O2 and H2O, BIP quality, Air Products) for 90 min and after that heating of sample was performed from 30 to 520 °C with the rate of 5 °C min−1. Carrier helium gas flow was set to 28 ml min−1. Hydrogen and diborane gas (B2H6) level was measured with the use of mass spectrometry by analyzing the intensity of ions with the ratio m/z = 2 (H2), 27 (B2H6) as well as 26 and 24 (species that may form owing to the decomposition of B2H6). For the purpose of graph plotting the measured pressure of escaping gases was normalized by the mass of the powder sample. Such a normalization allows a qualitative direct comparison of the peak intensities of various released gases observed on the mass spectroscopy (MS) plots that will be discussed in this paper. The mass normalizing was performed only to avoid misleading differences in signal intensities caused by different masses of the samples.
3. Results
3.1. Morphology of the powders before and after MCAS
Fig. 1 illustrates the SEM micrographs of the morphology of as-received TiF3 (Fig. 1a), graphene (Fig. 1b) and ultrafine filamentary carbonyl Ni (Fig. 1c and d) material, respectively. By comparing the SEM micrograph of TiF3 and that of the as-received LiBH4 (already reported in ref. 21 and 28), one can see that the TiF3 powder particles are finer than LiBH4, but more agglomerated. However, the graphene powder particles are very dispersed being in a platelet (flake) form, while the ultrafine filamentary carbonyl Ni powder exhibits agglomerated ball-like features (Fig. 1c) which exhibit submicrometric filamentary morphology under a high magnification (Fig. 1d). | ||
| Fig. 1 Scanning electron micrographs of as received constituent powders (a) TiF3, (b) graphene and (c and d) Ni. | ||
It is to be pointed out that the milling energy in the magneto-ball mill Uni-Ball-Mill 5 can be controlled by changing the number of hard steel balls in a milling vial and changing the angular positions of one or two strong NdFeB magnets.29 It has been demonstrated that the quantity of milling energy input, QTR(R) per unit mass hour (kJ g−1 h−1) which is injected into and stored in a milled powder for each particular milling mode with a fixed ball-to-powder mass ratio, R, in the magneto-ball mill Uni-Ball-Mill 5, can be calculated by a semi-empirical method.29 Thus, for the milling mode IMP68-4B-R132, which was applied in the present work, the milling energy input per hour (h) is QTR(132) = 72.8 kJ g−1 h−1 (Table 5 in ref. 29). Consequently, the total milling energy input per unit mass, QTR (kJ g−1), can be calculated for any milling duration in hours.
A dramatic refinement of the initial powder mixture after ball milling (BM), particularly in comparison to the large size of the as-received LiBH4 particulate, can be clearly seen in Fig. 2. However, some severely agglomerated powder particles are also observed after milling. In addition, there is no significant difference between the morphology of the ball milled powder without additives and the powder with the addition of 5 wt% Ni (Fig. 2e and f). However, a SEM micrograph of a sample with graphene (Fig. 2c and d) suggests less agglomeration when the milling time is increased from 1 to 5 h although this hypothesis would have to be quantitatively supported with detailed measurements of powder particle distribution.
 | ||
| Fig. 2 Scanning electron micrographs of ball milled (BM) powders for varying milling time (milling energy input). (a) (3LiBH4 + TiF3)-1 h BM, (b) (3LiBH4 + TiF3)-5 h BM (c) (3LiBH4 + TiF3) + 5 wt% graphene-1 h BM (d) (3LiBH4 + TiF3) + 5 wt% graphene-5 h BM (e) (3LiBH4 + TiF3) + 5 wt% Ni-1 h BM (f) (3LiBH4 + TiF3) + 5 wt% Ni-5 h BM. | ||
3.2. Mechanical dehydrogenation and microstructural evolution during ball milling (BM)
Substantial release of H2 during ball milling (BM) was already observed in the (2LiBH4 + FeCl2)20 and minimal in (nLiBH4 + MnCl2)21 systems. So far, the phenomenon of mechanical dehydrogenation for the (3LiBH4 + TiF3) system has never been reported in the literature. As shown in Fig. 3, the additive-free sample mechanically dehydrogenated 1.35 and 1.58 wt% H2 after milling with an energy input QTR = 72.8 kJ g−1 (1 h BM) and QTR = 364 kJ g−1 (5 h BM), respectively, while the sample with ultrafine Ni mechanically dehydrogenated 1.42 and 1.97 wt% H2 after 1 and 5 h BM, respectively. Apparently, regardless of the presence of ultrafine filamentary Ni both types of samples exhibit a similar mechanical dehydrogenation behavior during BM which shows that ultrafine Ni does not accelerate mechanical dehydrogenation rate to any measurable extent in contrast to its strong effect on the thermal dehydrogenation of hydrides containing submicrometric/nanometric Ni.2,3 | ||
| Fig. 3 The quantity of H2 desorbed during milling of the (3LiBH4 + TiF3) powder mixture without and with additives. | ||
On the other hand, a surprising aspect of mechanical dehydrogenation of the sample with graphene is that it showed less dehydrogenation up to QTR = 72.8 kJ g−1 (1 h BM) in comparison with the other two systems. However, the rate of dehydrogenation increased rapidly after 1 h of milling and the quantity of desorbed H2 reached 2.38 wt% after the energy input reached QTR = 364 kJ g−1 (5 h BM) (Fig. 3). As mentioned in the Experimental section reduced graphene oxide employed in the present work contains ∼1 wt% H2. That quantity of H2 is, most likely, released when a large energy input of QTR = 364 kJ g−1 (5 h BM) is injected into the powder. In this context, it is to be pointed out that after longer milling duration reduced graphene oxide is transformed, at least, partially to a highly dispersed amorphous carbon which may additionally enhance the release of H2 from the original graphene structure.
The XRD patterns of the samples after 1 and 5 h BM and complete dehydrogenation at three different temperatures are presented in Fig. 4a and b, respectively. Fig. 4a shows the XRD patterns of the additive-free powder mixture milled with an energy input, QTR = 72.8 kJ g−1 (1 h BM) compared to those after thermal treatment. The diffraction peaks of LiBH4 and TiF3 can still be seen after 1 h BM (Fig. 4a). Fig. 4b shows that after 5 h BM, the peak intensities of TiF3 are much lower than those after 1 h BM, and the peaks of LiBH4 are barely discernible. A trace of LiF diffraction peaks as well as very weak peaks of elemental Ti after 1 and 5 h BM, in addition to the presence of TiF3 diffraction peaks, indicate that a mechano-chemical reaction between the reactants started during BM but the amount of energy was still insufficient to complete the reaction. Moreover, Fig. 4a and b show that after dehydrogenation up to 100 °C the peaks of TiF3 in both samples milled for 1 and 5 h are not visible anymore, whereas the intensity of the LiF peaks substantially increased. The peaks of Ti are clearly visible as well.
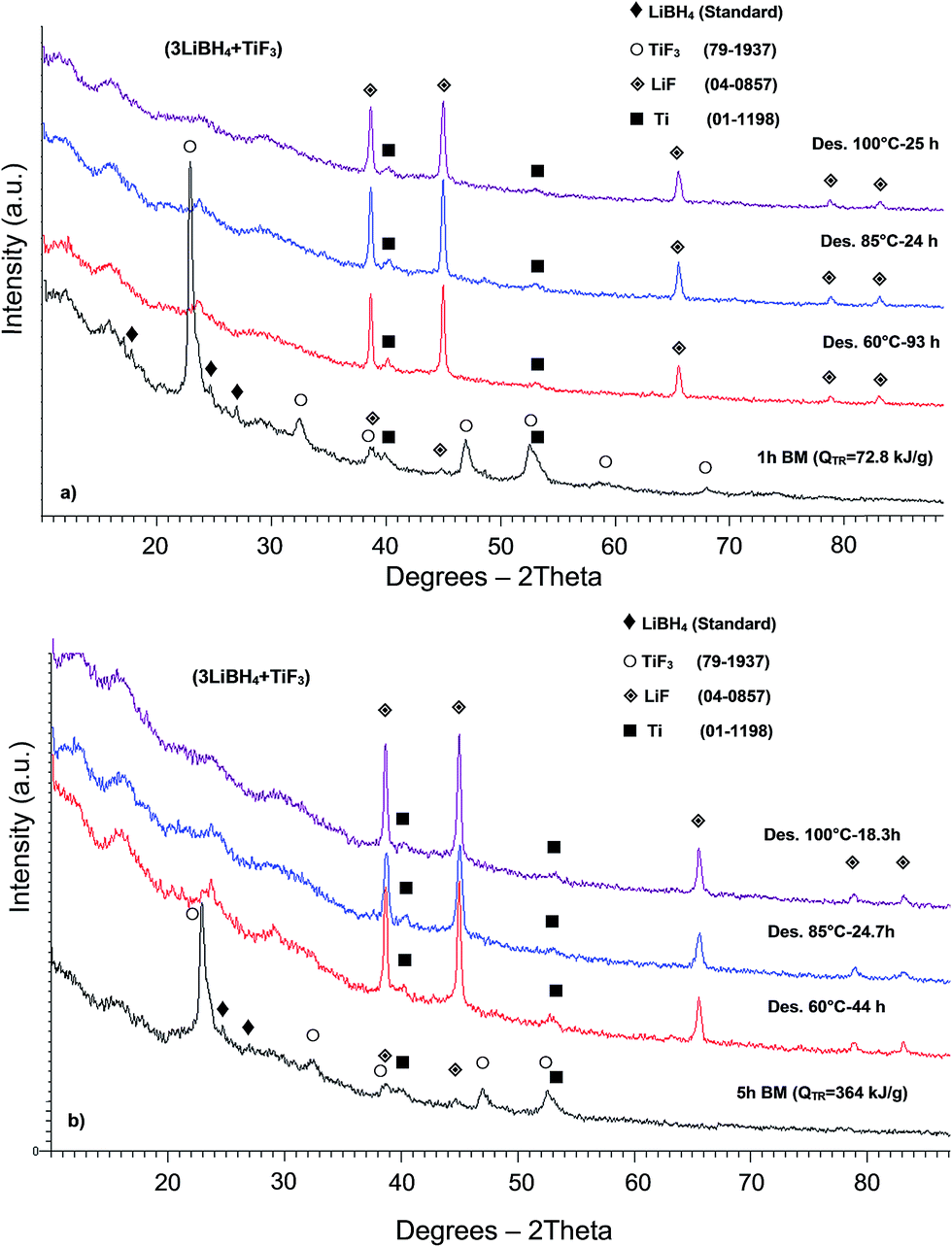 | ||
| Fig. 4 XRD patterns after ball milling (BM) and isothermal dehydrogenation at different temperatures for (a) (3LiBH4 + TiF3) with an energy input, QTR = 72.8 kJ g−1 (1 h BM) (b) (3LiBH4 + TiF3) with an energy input, QTR = 364 kJ g−1 (5 h BM). | ||
The XRD pattern of the powder mixture with 5 wt% graphene with an energy input, QTR = 72.8 kJ g−1 (1 h) and 364 kJ g−1 (5 h) are shown in Fig. 1S and 2S (ESI),† respectively. During BM, the diffraction peaks of LiBH4 nearly disappeared, whereas the TiF3 peaks are still visible. The LiF diffraction peaks which began to appear during BM becomes stronger after dehydrogenation at various temperatures (Fig. 1S and 2S†), while the lack of TiF3 peaks after thermal desorption indicates a similar phase transition as that for the additive-free samples.
Fig. 3S (ESI†) illustrates the XRD patterns of the sample with ultrafine Ni, ball milled with an energy input QTR = 72.8 kJ g−1 (1 h) compared to a sample after dehydrogenation at 60 °C. Diffraction peaks of Ni as well as TiF3, LiF and Ti are clearly seen after 1 h BM while dehydrogenation at 60 °C led to the disappearance of the TiF3 peaks. Similar behavior can be seen for the sample containing Ni after BM with an energy input QTR = 364 kJ g−1 (5 h BM) in Fig. 4S.†
The FT-IR spectrum for the additive-free sample, ball milled with an energy input QTR = 364 kJ g−1 which desorbed 1.58 wt% H2 during milling, is shown in Fig. 5a, and can be compared to the samples after isothermal dehydrogenation at 60 °C for 2.2 h (desorbed 1.58 wt% H2 during BM and additionally 0.99 wt% H2 during isothermal dehydrogenation) and 100 °C for 18.3 h (desorbed 1.58 wt% H2 during BM and additionally 4.8 wt% H2 during isothermal dehydrogenation) in Fig. 5b and c, respectively. Fig. 5d shows the FT-IR spectrum of pure LiBH4 which is plotted based on the data adapted from ref. 30.
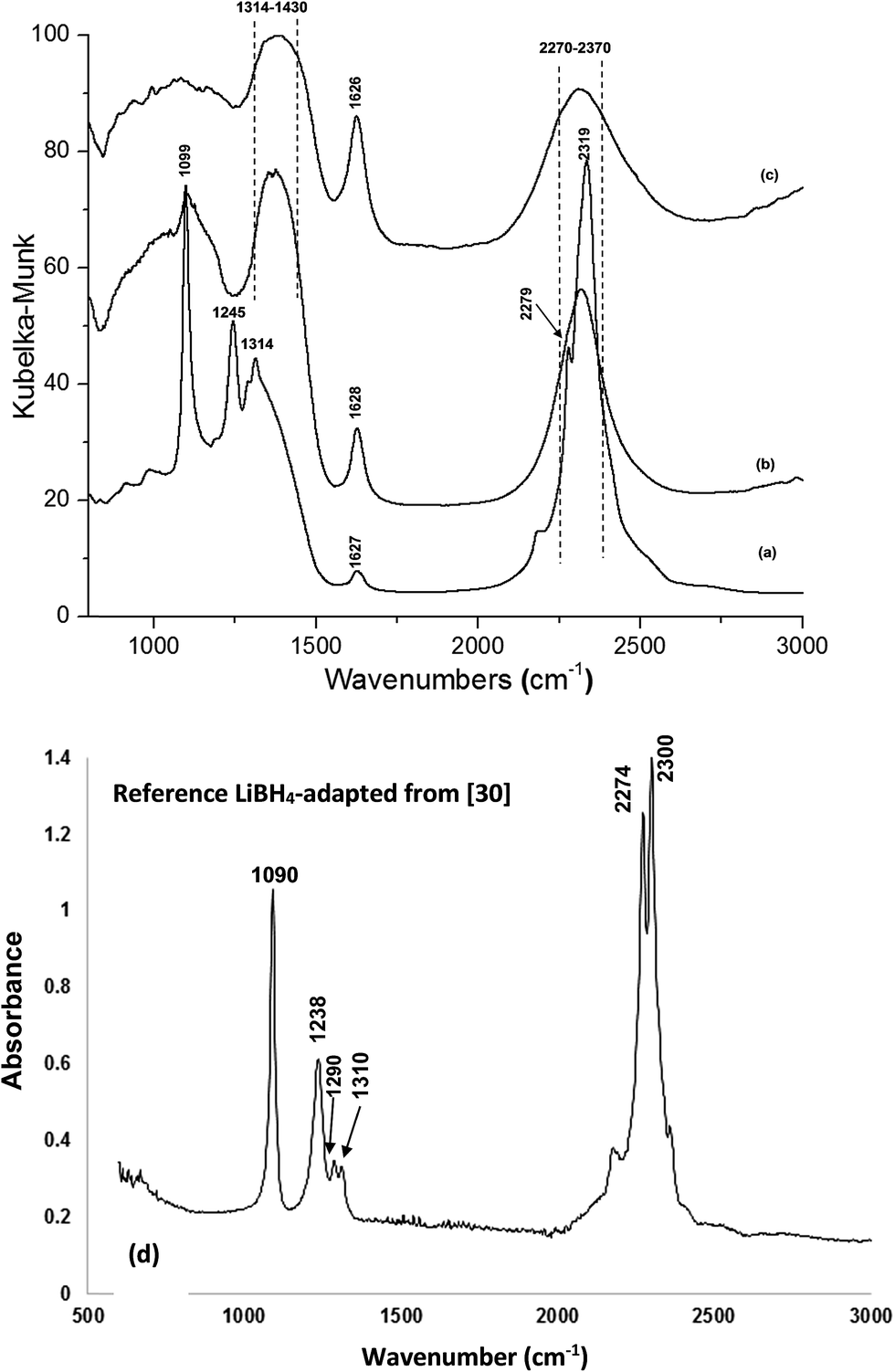 | ||
| Fig. 5 (a) FT-IR spectrum for the sample without additive with an energy input of QTR = 364 kJ g−1, 5 h BM (desorbed 1.58 wt% H2), (b) after 5 h BM and isothermal dehydrogenation at 60 °C for 2.2 h (desorbed 2.57 wt% H2), (c) after 5 h BM and isothermal dehydrogenation at 100 °C for 18.3 h (desorbed 6.38 wt% H2) and (d) pure LiBH4 adapted from ref. 30. | ||
3.3. Thermal behavior in DSC/TGA and mass spectrometry (MS)
Gas mass spectrometry (MS) as well as DSC/TGA results during temperature programmed desorption (TPD) up to 500 °C of the 1 h ball milled additive-free samples are shown in Fig. 6a and b and can be compared to those after 5 h ball milled in Fig. 6c and d, respectively. For the sample milled with an energy input QTR = 72.8 kJ g−1 (1 h BM), the release of hydrogen starts from about 50 °C as a principal gas with a maximum intensity around 120–140 °C and is accompanied by a miniscule quantity of diborane B2H6 (Fig. 6a). The ratio of H2 to B2H6 is 6015 up to 200 °C which increases to 6089 with temperature increasing to 500 °C. It is also worth noting that the release of B2H6 in the 1 h BM sample starts around 110 °C, with a maximum intensity at around 125–135 °C, indicating that B2H6 may not be released at low temperatures < 100 °C which strongly suggests that gas desorbed during ball milling (Fig. 3) is, indeed, pure H2.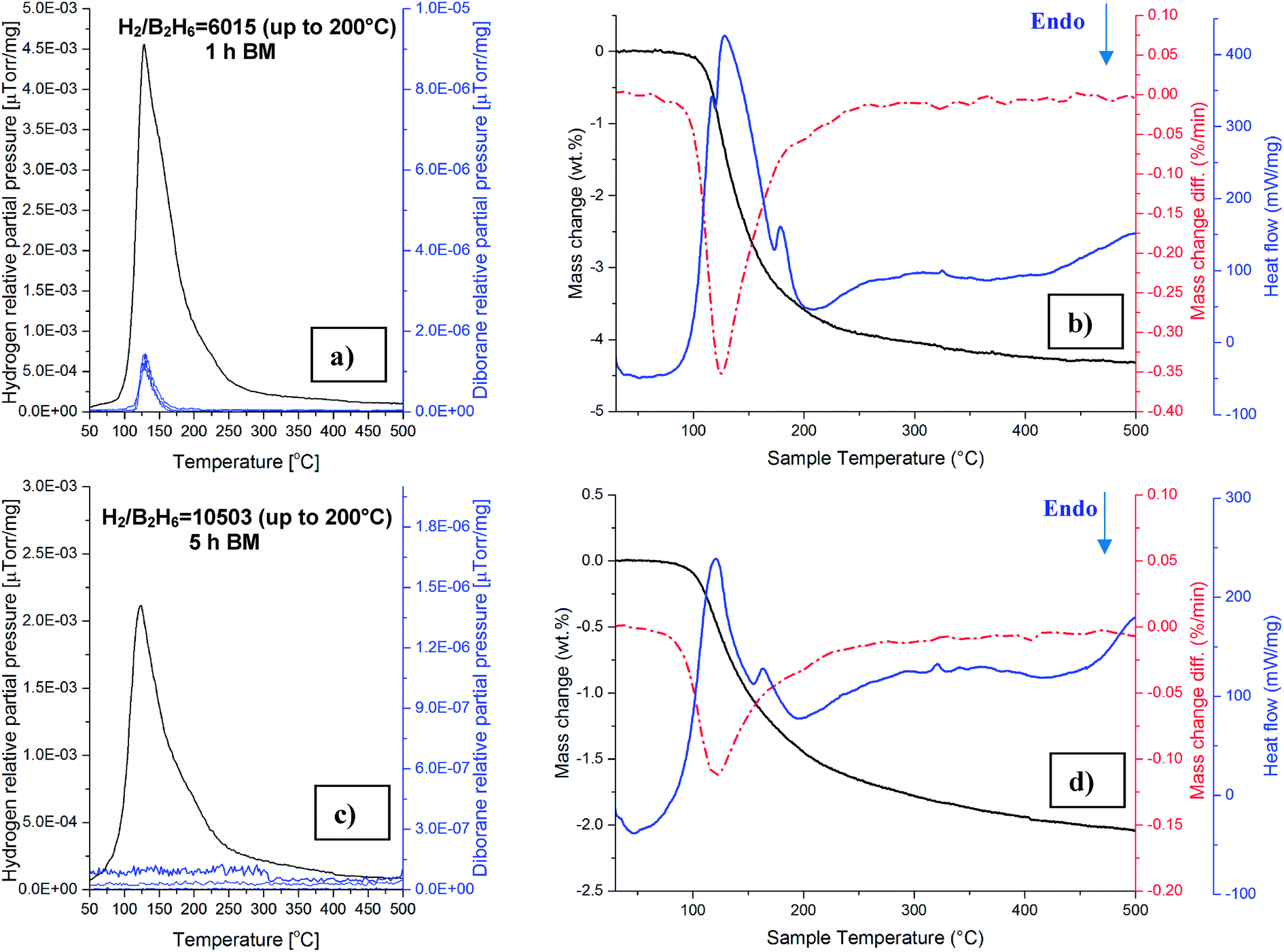 | ||
| Fig. 6 (a) Mass spectrometry (MS) gas desorption spectra for a (3LiBH4 + TiF3) sample ball milled for 1 h and (b) TG and DSC curves. (c) Mass spectrometry (MS) gas desorption spectra for a (3LiBH4 + TiF3) sample ball milled for 5 h and (d) TG and DSC curves. Heating rate 5 °C min−1. | ||
A very interesting finding is that, in contrast to 1 h BM sample, gas mass spectrometry of the sample after 5 h BM (QTR = 364 kJ g−1) does not show, within a resolution range of the MS instrument, any visible release of B2H6 which is also reflected in a huge intensity ratio H2/B2H6 = 10503 (Fig. 6c). The DSC curve of the 1 h BM sample (Fig. 6b) exhibits a large exothermic peak at around 125 °C and a very small overlapped exothermic peak at about 180 °C, whereas for the 5 h BM sample, the maxima of the first and second overlapped exothermic peaks occur at 120 °C and 170 °C (Fig. 6d), respectively. The TGA mass loss up to 200 °C was reduced from 3.58% to 1.44% by increasing the milling energy from 72.8 kJ g−1 (1 h) to 364 kJ g−1 (5 h) (compare Fig. 6b and d).
3.4. Isothermal dehydrogenation
Fig. 7 shows the isothermal dehydrogenation curves at varying temperatures from 60 to 200 °C for the additive-free samples (Fig. 7a and b), those with ultrafine filamentary Ni (Fig. 7c and d) and graphene (Fig. 7e and f) ball milled with energy input QTR = 72.8 and 364 kJ g−1. The quantities of H2 desorbed during BM and subsequent isothermal dehydrogenation at varying temperatures are summarized in Table 1S (ESI).† | ||
| Fig. 7 Dehydrogenation curves for (a) (3LiBH4 + TiF3)-1 h BM, (b) (3LiBH4 + TiF3)-5 h BM (c) (3LiBH4 + TiF3) + 5 wt% Ni-1 h BM (d) (3LiBH4 + TiF3) + 5 wt% Ni-5 h BM (e) (3LiBH4 + TiF3) + 5 wt% graphene-1 h BM (f) (3LiBH4 + TiF3) + 5 wt% graphene-5 h BM. | ||
As can be seen, in Table S1† for the 1 h BM additive-free sample, the total quantity of H2 desorbed at 60 °C within 93 h is 4.52 wt%, which increases to 5.81 wt% at 200 °C within 21 h. On the other hand, the same powder milled for 5 h (Fig. 7b) desorbed slightly less hydrogen (5.58 wt%) at 200 °C in comparison with the powder milled for 1 h owing to higher release of H2 during BM. The total quantities of H2 desorbed during BM and subsequent isothermal dehydrogenation at 200 °C for both the 1 and 5 h BM samples are same (7.16 wt%).
As mentioned in Section 3.2 (Fig. 3), the sample with 5 wt% Ni tends to release more H2 during BM than the additive-free one (column two in Table 1S†). It also shows a faster dehydrogenation rate than the additive-free sample during isothermal dehydrogenation (Fig. 7c). The time required for desorbing 4.00 wt% H2 for the sample with 5 wt% Ni after 1 h BM is 21.8 h at 60 °C, in contrast to 30 h for the additive-free sample at 60 °C.
The sample with 5 wt% graphene milled for 1 h (QTR = 72.8 kJ g−1) shows a slower thermal dehydrogenation rate at 60 °C than all other samples, and does not even reach 4.00 wt% H2 after 43 h thermal treatment (Fig. 7e). For the 5 wt% graphene sample ball milled for 5 h (QTR = 364 kJ g−1), the initial H2 desorption rate is quite rapid (Fig. 7f). However, after a short while the desorption starts saturating and the quantity of H2 does not exceed 3 wt% for any dehydrogenation temperature from 60 to 200 °C up to 24 h duration (Fig. 7f and Table 1S†). In addition, for that sample the total H2 quantity of 5.25 wt% desorbed during ball milling and at 200 °C (24 h) is the smallest one which is partially related to the fact that it exhibited the highest amount of desorption during ball milling among all the samples (Fig. 3 and Table 1S†) and also related to a rapid saturation of desorption rate at 200 °C (Fig. 7f). As mentioned earlier, after milling for 5 h (QTR = 364 kJ g−1) the reduced graphene oxide additive is to a large extent reduced to highly dispersed amorphous carbon. The presence of amorphous carbon can be responsible for a dehydrogenation behavior observed in Fig. 7f.
The apparent activation energy for dehydrogenation is estimated using the linear section of the dehydrogenation curves in the range of 60–120 °C in Fig. 7 and illustrated in the bar graph shown in Fig. 8. The apparent activation energy for thermal dehydrogenation for the additive-free sample slightly decreases with increasing milling energy from 72.8 kJ g−1 (1 h BM) to 364 kJ g−1 (5 h BM). The addition of 5 wt% Ni reduces the activation energy for the sample with a 72.8 kJ g−1 milling energy input (1 h BM) by comparison to the additive-free sample ball milled with the same energy input (1 h BM). Further milling of Ni containing sample for 5 h (QTR = 364 kJ g−1) leads to a slight increase in the apparent activation energy. The highest apparent energy for dehydrogenation is exhibited by a sample with 5 wt% graphene after injection of 72.8 kJ g−1 milling energy (1 h BM). However, as shown in Fig. 8, after 5 h BM (QTR = 364 kJ g−1) the apparent activation energy of the sample with 5 wt% graphene is dramatically reduced to about half of its value observed after 1 h BM (QTR = 72.8 kJ g−1), i.e. 42.9 ± 3.8 vs. 95.2 ± 1.9, respectively. It is most likely that this low apparent activation energy for the initial linear desorption period is related to a transformation of initial graphene into highly dispersed amorphous carbon. There are some observations published in the literature that ball milling of borohydrides, like LiBH4, with carbonaceous additives substantially improves their dehydrogenation kinetics.31–34 The explanations suggested for the effect of carbonaceous species on the enhancement of kinetics vary from the formation of Li2C2 during dehydrogenation,34 through a synergy of catalytic and good thermal conductivity effects31 to heterogeneous nucleation and micro-confinement effects.32,33 However, as can be seen in Fig. 7f, simultaneously, the dispersed amorphous carbon leads to a quick saturation of desorption manifested in a flattening of a desorption curve. This peculiar behavior may indicate a reaction between carbon and other species which rapidly exhausts its catalytic effect. Further studies are required to elucidate the cause of that peculiar desorption behavior in Fig. 7f.
 | ||
| Fig. 8 Apparent activation energy (kJ mol−1) for dehydrogenation for the samples with and without additives after 1 and 5 h BM. | ||
In order to gain the insight about the size of the phases formed during dehydrogenation the crystallite size of LiF and the lattice strain after isothermal dehydrogenation at 60 °C for a specific time duration are listed in Table 1. Apparently, LiF is nanometric.
| Sample | Crystallite size LiF (nm) | Lattice strain LiF (%) | Time of desorption at 60 °C (h) |
|---|---|---|---|
| (3LiBH4 + TiF3)-1 h BM | 33.5 | 0.81 | 93.3 |
| (3LiBH4 + TiF3)-5 h BM | 49.5 | 0.22 | 44.0 |
| (3LiBH4 + TiF3) + 5 wt% Ni-1 h BM | 35.2 | 1.01 | 44.7 |
| (3LiBH4 + TiF3) + 5 wt% Ni-5 h BM | 36.5 | 1.09 | 49.5 |
| (3LiBH4 + TiF3) + 5 wt% graphene-1 h BM | 34.7 | 0.96 | 47.1 |
| (3LiBH4 + TiF3) + 5 wt% graphene-5 h BM | 25.2 | 1.35 | 39.4 |
Fig. 9 shows dehydrogenation curves obtained at 100 °C for the additive-free sample ball milled with an energy input, QTR = 72.8 kJ g−1 (1 h) and 364 kJ g−1 (5 h) and subsequently stored for 7 and 3 month duration, respectively, at room temperature under a slight overpressure of argon. Fig. 9a shows that the quantity of H2 desorbed from the sample ball milled with an energy input, QTR = 72.8 kJ g−1 (1 h), is 5.2 wt% H2 within 24 h, whereas at the same desorption time for the sample stored for 7 months it decreases to 2.8 wt% H2. Fig. 9b illustrates the reduction in the quantity of H2 desorbed from the sample ball milled with an energy input 364 kJ g−1 (5 h) from 5.00 wt% to 1.72 wt% in 42.5 h during 3 months of storage.
 | ||
| Fig. 9 Dehydrogenation curves for 3LiBH4 + TiF3 at 100 °C for (a) BM for 1 h and stored for 7 months and (b) BM for 5 h and stored for 3 months. | ||
4. Discussion
As mentioned earlier, very little is found in the literature on the subject of the mechanical and thermal dehydrogenation behavior of the LiBH4–TiF3 system. Table 2 summarizes the milling conditions, dehydrogenation temperature and phase analysis after BM for the LiBH4–TiF3 system with varying molar ratios and with additives. Recently, Fang et al.17 claimed in situ formation and decomposition of amorphous Ti(BH4)3 obtained from MCAS (Table 2). The XRD pattern of the BM mixture exhibited only the Bragg peaks of TiF3, which suggested that no mechano-chemical reaction occurred during milling. The authors reported no H2 release during ball milling which is contradictory to the observations of H2 release in this work (Fig. 3). Within the temperature range 70–90 °C, the authors observed about 5.0–5.6 wt% H2 release from the ball milled sample with an estimated apparent activation energy of ∼19 kJ mol−1 which seems to be unusually low. Differential scanning calorimetry (DSC) showed a strongly exothermic peak centered at around 90 °C, accompanied by 5.7 wt% mass loss obtained from thermogravimetric analysis (TGA). No diborane (B2H6) gas was detected by mass spectrometry (MS) during dehydrogenation in DSC/TGA, in contrast to this work and the report by Soloveichik11 suggesting formation of B2H6. The XRD patterns after dehydrogenation exhibited only LiF diffraction peaks. The authors proposed that Ti(BH4)3 was formed at slightly elevated temperatures and decomposed according to the following reaction:17| 3LiBH4 + TiF3 (BM) → (thermal) Ti(BH4)3 + 3LiF → 3LiF + 3B + 5H2 + TiH2 | (3) |
| Ref. | System | BM | Phases after BM from XRD | Heating (°C) | Phases after heating from XRD/XPS |
|---|---|---|---|---|---|
| 17 | 3LiBH4 + TiF3 | (2 h) Fritch 7-Planetary 500 rpm | TiF3 | 70–90 | LiF/XPS: LiF + B + TiH2 |
| 18 | 3LiBH4 + TiF3 | (15 min) Planetary Qm-isp2 | TiF3 + LiBH4 | 600 | LiF/XPS: LiF + TiB2 + B |
| 18 | 50LiBH4 + TiF3 | (15 min) Planetary Qm-isp2 | TiF3 + LiBH4 | 600 | LiF + LiH |
| 19 | LiBH4 + 20 wt% Fe2O3 + 30 wt% TiF3 | (5 h) 350 rpm Qm-3sp2 | TiF3 + LiBH4 + Fe2O3 | 100 | TiF3 + LiBH4 + Fe2O3 + LiF |
| 14 | LiBH4 + TiF3 + 20 wt% SiO2 | (1 h) 450 rpm Planetary Qm-isp | LiF + TiB2 | 70 | LiF + TiB2 |
| This work | (3LiBH4 + TiF3) | (1 h) 200 rpm magneto-mill | TiF3 + LiBH4 + LiF + Ti | 60 | LiF + Ti |
| This work | (3LiBH4 + TiF3) | (5 h) 200 rpm magneto-mill | TiF3 + LiF + Ti | 60 | LiF + Ti |
The theoretical capacity of this reaction is 5.9 wt% H2, which agrees with the slightly lower observed H2 release of ∼5.0–5.6 wt%. However, no TiH2 diffraction peaks were detected in the XRD patterns after dehydrogenation, as required by reaction (3) although they were evidenced by XPS. Furthermore, one can calculate the standard enthalpy (heat) of reaction (3), ΔH0(reaction), given as the difference between the standard enthalpies (heats) of formation of the products and the reactants35 in the following form:
| ΔH0(reaction) = ∑ΔH0f(products) − ∑ΔH0f(reactants) | (4) |
| ΔH0(reaction (3)) = [3 × (−616.93) + (3 × 0) + (5 × 0) − 142.39] − [3 × (−190.46) + (−1435.11)] = +13.31 kJ mol−1 or +2.66 kJ mol−1 H2. | (5) |
This estimate would suggest that the thermal dehydrogenation reaction (3) should have a weakly endothermic character, which is in a contradiction to the exothermic reaction character observed in DSC by Fang et al.17 However, Arita et al.38 reported that ΔH0f(TiH2) = −179 kJ mol−1, which would make the standard heat of the reaction (3) very weakly exothermic, with ΔH0(reaction (3)) = −23.3 kJ mol−1 or −4.7 kJ per mol H2. Therefore, such a large exothermic DSC peak in ref. 17 still remains to be clearly explained.
More recently, Guo et al.18 ball milled a (3LiBH4–TiF3) mixture in a planetary ball mill under argon for 15 min. The XRD patterns of the BM mixtures exhibited only the Bragg peaks of LiBH4 and TiF3, indicating that no reaction occurred during BM. Their finding is contradictory to the present results showing clearly the presence of the LiF and Ti diffraction peaks (Fig. 4 and 1S–4S†). After dehydrogenation at 600 °C, the XRD patterns exhibited only the LiF Bragg diffraction peaks, indicating that some chemical reaction occurred during thermolysis. The X-ray photo electron spectroscopy (XPS) analysis of the samples dehydrogenated at 350 and 600 °C showed some spectra peaks, interpreted by the authors as corresponding to TiB2, TiO2 and B2O2. The authors explained that the presence of oxides was a result of exposure to air when the samples were loaded into the XPS apparatus. The lack of the TiB2 peaks on the XRD patterns was explained by the amorphous structure of TiB2. On the basis of their results, the following thermal reaction path was proposed:18
| 3LiBH4 + TiF3 → 3LiF + TiB2 + B + 6H2 | (6) |
The theoretical H2 capacity of reaction (6) is 7.12 wt%. One can also estimate the standard enthalpy of reaction (6), ΔH0(reaction (6)), taking standard molar enthalpies for LiBH4, TiF3 and LiF as those in eqn (3) and that for TiB2 as equal to −280.33 kJ mol−1,36 which gives ΔH0(reaction (6)) = −124.6 kJ mol−1 or −20.77 kJ mol−1 H2. A negative standard enthalpy (heat) of reaction (6) indicates an exothermic nature of the reaction, which was, indeed, observed by DSC in ref. 18. Finally, it must be pointed out that Guo et al.18 did not investigate the isothermal dehydrogenation behavior of the BM mixtures as was done in ref. 17. Interestingly, reaction (6) is in agreement with a claim by Soloveichik,11 who suggested the formation of TiB2 upon decomposition of Ti(BH4)3, but it does not release B2H6 which Soloveichik11 also claimed to occur and what is observed in this work.
One of the interesting findings in our work is the observed release of H2 during ball milling (Fig. 3). This clearly shows that mechano-chemical reaction must have occurred during ball milling. This is supported by the presence of LiF and Ti diffraction peaks after BM which strongly support the occurrence of mechano-chemical which, so far, has never been reported in the literature.
Furthermore, this work clearly shows that the release of B2H6 during thermal dehydrogenation depends on the quantity of milling (mechanical) energy injected into the powder mixture. A thermal release of small quantities of B2H6 is observed for the sample milled with the energy input QTR = 72.8 kJ g−1 (1 h BM) at a very large ratio of H2/B2H6 = 6015 (Fig. 6a). In contrast, B2H6 is not observed for the 5 h BM sample (QTR = 364 kJ g−1) (Fig. 6c). The FT-IR measurements shown in Fig. 5a for the 5 h BM sample confirm the existence of LiBH4 which exhibits barely discernible XRD peaks in Fig. 4b. As can be seen in Fig. 5d, the FT-IR of pure LiBH4 adapted from the data base of borohydrides presented on ref. 30, strongly supports the existence of LiBH4 after 5 h BM. Two IR active modes of bending in the range of 1050–1430 cm−1 and stretching in the range of 2270–2370 cm−1 for LiBH4 (tetrahedral bond)20,30,39 are obvious in the FT-IR spectrum of the sample after 5 h BM. However, the other peak within the range of 1600–1650 cm−1 may be related a trace of moisture present in the glove box during loading our sample into a FT-IR apparatus.40 Interestingly, the intensity of characteristic peaks of LiBH4 at 1050–1430 cm−1 and 2270–2370 cm−1 gradually decreased and their width increased with dehydrogenation of the milled powder. Isothermal dehydrogenation at 60 °C for 2.2 h caused a disappearance of one of the IR peaks in the bending mode (1245 cm−1). The peaks at 1099 cm−1 became wider while that at 1314 cm−1 shifted to the higher wavelength. Dehydrogenation at 100 °C for 18.3 h (Fig. 5c) shows a complete disappearance of the first two peaks of B–H bending mode (1099 and 1245 cm−1) and the width broadening of shifted peak at 1314–1430 cm−1. A comparison of FT-IR spectra of the ball milled sample (Fig. 5a) with the dehydrogenated ones (Fig. 5b and c) indicates that the mechano-chemical reaction started during BM but the amount of energy was insufficient to complete the reaction. However, according to the phase identification by XRD, the presence of LiF and Ti diffraction peaks which appeared after BM as well as after thermal dehydrogenation indicates that TiF3 reacts with LiBH4 and, most likely, forms Ti(BH4)3. These results are in accord with the FT-IR study done by Sun et al.,41 who investigated the formation of Ti(BH4)3 in the (3LiBH4 + TiCl3) powder mixture during milling. They claimed that Ti(BH4)3 was very unstable at the ambient environment and decomposed rapidly to TiH2, B and H2. They only observed the signature bands of B–H in LiBH4 using FT-IR while they could not detect LiBH4 by XRD. Au et al.42 reported that a possible reason of the LiBH4 peaks disappearing during BM with halides could be due to either the reaction of LiBH4 with a halide (in their case: MgCl2 and TiCl3) resulting in its decomposition even before heating or transformation of LiBH4 to an amorphous state or both of the above. In all previous studies, it was proposed that a metal borohydride Ti(BH4)3 decomposed at RT due to its unstable structure although no direct evidence in support of the existence of Ti(BH4)3 after MCAS of LiBH4 and TiCl3 (ref. 13, 41 and 43) has ever been provided. A similar behavior was also reported in the milling of LiBH4 with FeCl2, in which LiBH4 was not observed by XRD while it was observed in an FT-IR spectrum.20 It was proposed that unstable Fe(BH4)2 decomposed very fast and that was why possibly amorphous LiBH4 was the only phase detected by the FT-IR. It seems that the new unstable borohydrides form and decompose in stepwise reactions of LiBH4 with different halides (TiF3, TiCl3, FeCl2).
It is clearly seen in Fig. 6 that in the sample ball milled for 1 h the release of B2H6 starts around 100–110 °C which implicates that B2H6 is unlikely to be released at temperatures < 100 °C. That strongly suggests that gas desorbed during BM was pure H2. Therefore, assuming that, indeed, unstable Ti(BH4)3 is being formed, the first possible reaction formation/decomposition during ball milling could be as follows where Ti(BH4)3 forms and simultaneously rapidly decomposes:
 | (7) |
The theoretical capacity of this reaction is 7.1 wt% H2 which is much larger than even the observed 2.38 wt% H2, released as a result of mechanical dehydrogenation for 5 h in the presence of graphene which apparently moderately accelerates mechanical dehydrogenation rate as discussed earlier. The milling energy input of 364 kJ g−1 is apparently still insufficient to mechanically dehydrogenate even barely 50% of the theoretical capacity of reaction (7). In contrast to a few other complex hydride systems such as LiAlH4/MnCl2 and LiAlH4/LiNH2 (ref. 3 and 5) and LiBH4/FeCl2,20 capable of mechanical dehydrogenation of 4–5 wt% H2, the present system LiBH4/TiF3 seems to be moderately resistant to mechanical dehydrogenation (Table 1S†) even with envisaged catalytic additives like Ni and graphene. However, to put it into perspective the present hydride system is more prone to mechanical dehydrogenation than the LiBH4/MnCl2 system which mechanically dehydrogenated barely 0.7 wt% H2 after 5 h BM.21
As shown in Fig. 6, the release of diborane gas, B2H6, is observed for the 1 h BM sample but not observed for the 5 h BM sample. Therefore, three alternative, general reactions could be proposed for the thermal decomposition of the ball milled (3LiBH4 + TiF3) system:
 | (8) |
 | (9) |
 | (10) |
Reaction (9) contains TiH2 after decomposition. It was reported in ref. 44 that TiH2 if present in such reaction would have to decompose with two endothermic peaks at around 460 and 560 °C. However, no endothermic peaks are observed in our DSC curves (Fig. 6b and d). Standard enthalpy of reaction (9), ΔH0(reaction (9)), can be estimated as above taking standard molar enthalpies for LiBH4, TiF3, LiF and TiH2 as those in eqn (2) and that for B2H6 in eqn (7) which gives ΔH0(reaction (9)) = +11.35 and +46.91 kJ mol−1 for 0 and 1B2H6, respectively.
The standard enthalpy of reaction (10), ΔH0(reaction (10)), varies from −124.63 to +51.10 kJ mol−1 for 0B2H6 and 1B2H6, respectively. In other words, reaction (10) has an exothermic character whenever number of B2H6 moles (y) is not larger than 0.83, which agrees with the exothermic character of the DSC curves in Fig. 6. However, the problem is that eqn (10) requires the formation of TiB2 which is not observed on the pertinent XRD patterns after dehydrogenation of samples without additives (Fig. 4) as well those with catalytic additives (Fig. 1S–4S†). If one assumes that TiB2 is amorphous, as was also suggested by Guo et al.,18 and Ti in reaction (10) is nanocrystalline then this reaction agrees reasonably with the experimental results obtained in this work.
Finally, it is to be pointed out that it is observed in Fig. 9 that the sample ball milled with an energy input QTR = 72.8 kJ g−1 (1 h BM) desorbed after 7 months of storage about 2.4 wt% H2 while the sample milled with an energy input QTR = 364 kJ g−1 (5 h BM) desorbed after 3 months of storage about 3.28 wt% H2. In comparison with a number of ball milled nanocomposites, particularly those based on LiAlH4, containing catalytic additives,3 the present borohydride system exhibits a comparable dehydrogenation rate during a long-term storage at room temperature. However, comparing with other borohydride systems, a nanocrystalline Mn(BH4)2 hydride released only a small amount of about 0.5 wt% H2 within 80 days and subsequently stabilized up to 120 days of further storage.21 As reviewed in ref. 3 and 5 there are a number of possible practical engineering applications for a long-duration hydrogen discharge for generating quantities of hydrogen on demand for auxiliary power generation systems coupled with PEM fuel cells and batteries such as low power remote fuel cells, portable gas analyzers and smartphones.3,5,45 These materials can also be used in a number of chemical processes where a continuously reducing atmosphere is needed for a completion of the process.3,5 They could also have an application in a military sector for cartridges supplying hydrogen to micro fuel cells in portable devices needed for soldiers on a mission in remote areas.46
5. Conclusions
(1) During ball milling (BM) of the (3LiBH4 + TiF3) system a mechano-chemical reaction starts occurring between LiBH4 and TiF3 after injecting the energy input QTR = 72.8 kJ g−1 (1 h BM) which accelerates with increasing milling time to 5 h (QTR = 364 kJ g−1).(2) X-ray diffraction (XRD) shows the new phases LiF and Ti present in the microstructure accompanied by retained TiF3 and LiBH4.
(3) A mechanical dehydrogenation phenomenon occurs during mechano-chemical reaction resulting in a release of hydrogen.
(4) The ultrafine filamentary carbonyl Ni additive does not measurably accelerate the rate of mechanical dehydrogenation up to 5 h BM (QTR = 364 kJ g−1).
(5) The presence of graphene (reduced graphene oxide) does not accelerate the rate of mechanical dehydrogenation up to 1 h BM (QTR = 72.8 kJ g−1) but then the quantity of released H2 dramatically increases up to 5 h BM (QTR = 364 kJ g−1) with the quantity of 2.38 wt% H2 finally desorbed, most likely, due to an additional release of H2 contained in graphene.
(6) The ball milled samples are capable of desorbing H2 at a very low temperature of 60 °C resulting in the release of 4 wt% H2 within 33.4 h (4.52 wt% H2 within 93 h). The average apparent activation energy for thermal dehydrogenation equals 88.2 ± 0.8 and 81.8 ± 4.7 kJ mol−1 for samples ball milled with energy input QTR = 72.8 kJ g−1 (1 h BM) and QTR = 364 kJ g−1 (5 h BM), respectively.
(7) The addition of 5 wt% ultrafine Ni mildly reduces the average activation energy to 79.7 ± 4.0 kJ mol−1 for sample milled with a QTR = 72.8 kJ g−1 (1 h BM) but then slightly increases to 84.6 ± 1.3 kJ mol−1 after the energy input QTR = 364 kJ g−1 (5 h BM). The highest average apparent activation energy of 95.2 ± 1.9 kJ mol−1 is exhibited by a sample with 5 wt% graphene milled with QTR = 72.8 kJ g−1 (1 h BM) which is reduced to 42.9 ± 3.8 kJ mol−1 after the energy input QTR = 364 kJ g−1 (5 h BM).
(8) During thermal dehydrogenation the intensity of diffraction peaks of retained TiF3 and LiBH4 disappears while the intensity of LiF and Ti peaks dramatically increases which confirms the occurrence of a continuous thermally activated reaction between LiBH4 and TiF3 initiated during ball milling.
(9) Mass spectrometry shows that the principal gas released during thermal desorption is hydrogen although the sample milled with energy input QTR = 72.8 kJ g−1 (1 h BM) shows a miniscule quantity of diborane gas, B2H6, which starts around 110 °C, with a maximum intensity at around 125–135 °C. In contrast, the sample milled with energy input QTR = 364 kJ g−1 (5 h BM) does not show, within a detectability limit of the mass spectrometer, any release of B2H6. It clearly shows that the release of B2H6 during thermal dehydrogenation depends on the quantity of milling (mechanical) energy injected into the powder mixture.
(10) DSC measurements show exothermic peaks for both the 1 and 5 h BM samples regardless of milling energy input.
(11) The ball milled (3LiBH4 + TiF3) system is able to slowly discharge up to about 3 wt% H2 during a few months storage at room temperature.
Acknowledgements
This research was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant awarded to Prof R. A. Varin which is gratefully acknowledged. The authors are grateful to Prof. Linda Nazar from the Department of Chemistry, University of Waterloo, for allowing access to the XRD equipment. The authors are also grateful to Dr Steve Kornic from McMaster University for conducting the FT-IR analysis. The authors thank Dr John Shu from Cnem Corp. (Canada) for donating ultrafine filamentary carbonyl nickel (Ni) powders.References
- D. S. Scott, Smelling land-the hydrogen defense against climate catastrophe, Canadian Hydrogen Association, Westmount, QC, 2007 Search PubMed.
- R. A. Varin, T. Czujko and Z. S. Wronski, Nanomaterials for solid state hydrogen storage, New York, Springer Science Business Media, 2009 Search PubMed.
- R. A. Varin and Z. S. Wronski, Progress in hydrogen storage in complex hydrides, in Renewable Hydrogen Technologies. Production, Purification, Storage, Applications and Safety, ed. L. M. Gandia, G. Arzamendi and P. M. Diéguez, Elsevier, 2013, ch. 13, pp. 293–332, ISBN: 978-0-444-56352-1 Search PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, High capacity hydrogen storage materials: attributes for automotive applications and techniques for materials discovery, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- R. A. Varin and A. R. Shirani Bidabadi, Nanostructured, complex hydride systems for hydrogen generation, AIMS Energy, 2015, 3, 121–143 CrossRef.
- http://energy.gov/sites/prod/files/2014/03/f12/targets_onboard_hydro_storage.pdf; http://energy.gov/sites/prod/files/2015/01/f19/fcto_myrdd_table_onboard_h2_storage_systems_doe_targets_ldv.pdf.
- Toyota Hydrogen Fuel Cell Vehicles to be available in 2016, available from: http://www.hngn.com/articles/33584/20140612/toyota-hydrogen-fuel-cell-vehicles-to-be%20available-in-2016.htm, New version of the Mirai expected to be launched ahead of the 2020 Olympic Games, http://www.hydrogenfuelnews.com/toyota-plans-to-launch-a-new-version-of-its-fuel-cell-vehicle-in-2019/8528633/.
- Hyundai first to offer hydrogen fuel cell vehicles to Canadian public, available from: http://www.newswire.ca/en/story/1453489/hyundai-first-to-offer-hydrogen-fuel-cell-vehicles-to-canadian-public.
- H. R. Hoekstra and J. J. Katz, The preparation and properties of the group IV-B metal borohydrides, J. Am. Chem. Soc., 1949, 71, 2488–2492 CrossRef CAS.
- J. A. Jensen and G. S. Girolami, Transition metal tetrahydridoborates as models of methane activation: synthesis and structure of Ti(BH4)3(PMe3)2, J. Chem. Soc., Chem. Commun., 1986, 1160–1163 RSC.
- G. L. Soloveichik, Metal borohydrides as hydrogen storage materials, Mater. Matters, 2007, 2, 11–14 CAS.
- M. Au, A. Jurgensen and K. Zeigler, Modified lithium borohydrides for reversible hydrogen storage (2), J. Phys. Chem. B, 2006, 110, 7062–7067 CrossRef CAS PubMed.
- M. Au, A. R. Jurgensen, W. A. Spencer, D. L. Anton, F. E. Pinkerton, S.-J. Hwang, C. Kim and R. C. Bowman Jr, Stability and reversibility of lithium borohydrides doped by metal halides and hydrides, J. Phys. Chem. C, 2008, 112, 18661–18671 CAS.
- Y. Zhang, W.-S. Zhang, M.-Q. Fan, S.-S. Liu, H.-L. Chu, Y.-H. Zhang, X.-Y. Gao and L.-X. Sun, Enhanced hydrogen storage performance of LiBH4–SiO2–TiF3 composite, J. Phys. Chem. C, 2008, 112, 4005–4010 CAS.
- P. Wang, L. Ma, Z. Fang, X. Kang and P. Wang, Improved hydrogen storage property of Li–Mg–B–H system by milling with titanium trifluoride, Energy Environ. Sci., 2009, 2, 120–123 CAS.
- A. Züttel, S. Rentsch, P. Fischer, P. Wenger, P. Sudan, P. Mauron and C. Emmenegger, Hydrogen storage properties of LiBH4, J. Alloys Compd., 2003, 356–357, 515–520 CrossRef.
- Z. Z. Fang, L. P. Ma, X. D. Kang, P. J. Wang and H. M. Cheng, In situ formation and rapid decomposition of Ti(BH4)3 by mechanical milling LiBH4 with TiF3, Appl. Phys. Lett., 2009, 94, 044104 CrossRef.
- Y. H. Guo, X. B. Yu, L. Gao, G. L. Xia, Z. P. Guo and H. K. Liu, Significantly improved dehydrogenation of LiBH4 destabilized by TiF3, Energy Environ. Sci., 2010, 3, 465–470 CAS.
- H. Zhang, Z. Cao, L. Sun, Y. Sun, F. Xu, H. Liu, J. Zhang, Z. Huang, X. Jiang, Z. Li, S. Liu, S. Wang, C. Jiao, H. Zhou and Y. Sawada, Improved dehydrogenation/rehydrogenation performance of LiBH4 by doping mesoporous Fe2O3 or/and TiF3, J. Therm. Anal. Calorim., 2013, 112, 1407–1414 CrossRef CAS.
- R. A. Varin and A. R. Shirani Bidabadi, Rapid, ambient temperature hydrogen generation from the solid state Li–B–Fe–H system by mechano-chemical activation synthesis, J. Power Sources, 2015, 284, 554–565 CrossRef CAS.
- R. A. Varin and A. R. Shirani Bidabadi, The effect of milling energy input during mechano-chemical activation synthesis (MCAS) of the nanocrystalline manganese borohydride (Mn(BH4)2) on its thermal dehydrogenation properties, Int. J. Hydrogen Energy, 2014, 39, 11620–11632 CrossRef CAS.
- Y. Zhu, J. Zou and X. Zeng, Study on reversible hydrogen sorption behaviors of 3LiBH4/graphene and 3LiBH4/graphene–10 wt% CeF3 composites, RSC Adv., 2015, 5, 82916–82923 RSC.
- A. Calka and A. P. Radlinski, Universal high performance ball-milling device and its application for mechanical alloying, Mater. Sci. Eng., A, 1991, 134, 1350–1353 CrossRef.
- B. W. Ninham and A. Calka, WO9104810, 1991; B. W. Ninham and A. Calka, US5383615, 1995; B. W. Ninham and A. Calka, CA2066740, 1991; B. W. Ninham and A. Calka, EP0494899, 1992; B. W. Ninham and A. Calka, AU643949, 1990.
- R. A. Varin, S. Li, Z. Wronski, O. Morozova and T. Khomenko, The effect of sequential and continuous high-energy impact mode on the mechano-chemical synthesis of nanostructured complex hydride Mg2FeH6, J. Alloys Compd., 2005, 390, 282–296 CrossRef CAS.
- G. Sandrock, K. Gross, G. Thomas, C. Jansen, D. Meeker and S. Takara, Engineering consideration in the use of catalyzed sodium alanates for hydrogen storage, J. Alloys Compd., 2002, 330–332, 696–701 CrossRef CAS.
- G. K. Williamson and W. H. Hall, X-ray line broadening from filed aluminum and wolfram, Acta Metall., 1953, 1, 22–31 CrossRef CAS.
- A. R Shirani Bidabadi, A. Korinek, G. A. Botton and R. A. Varin, High resolution transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDS) and X-ray diffraction studies of nanocrystalline manganese borohydride (Mn(BH4)2) after mechano-chemical synthesis and thermal dehydrogenation, Acta Mater., 2015, 100, 392–400 CrossRef.
- R. Parviz and R. A. Varin, Combined effects of molar ratio and ball milling energy on the phase transformations and mechanical dehydrogenation in the lithium amide-magnesium hydride (LiNH2 + nMgH2)(n = 0.5–2.0) nanocomposites, Int. J. Hydrogen Energy, 2013, 38, 8213–8327 CrossRef.
- Université de Genève, http://www.unige.ch/sciences/chifi/?ftirdb.html.
- S. Thiangviriya and R. Utke, LiBH4 nanoconfined in activated carbon nanofiber for reversible hydrogen storage, Int. J. Hydrogen Energy, 2015, 40, 4167–4174 CrossRef CAS.
- Z.-Z. Fang, X.-D. Kang and P. Wang, Improved hydrogen storage properties of LiBH4 by mechanical milling with various carbon additives, Int. J. Hydrogen Energy, 2010, 35, 8247–8252 CrossRef CAS.
- Z. Z. Fang, P. Wang, T. E. Rufford, X. D. Kang, G. Q. Lu and H. M. Cheng, Kinetic- and thermodynamic-based improvements of lithium borohydride incorporated into activated carbon, Acta Mater., 2008, 56, 6257–6263 CrossRef CAS.
- X. B. Yu, Z. Wu, Q. R. Chen, Z. L. Li, B. C. Weng and T. S. Huang, Improved hydrogen storage properties of LiBH4 destabilized by carbon, Appl. Phys. Lett., 2007, 90, 034106 CrossRef.
- http://chemwiki.ucdavis.edu/Physical_Chemistry/Thermodynamics/State_Functions/Enthalpy/Standard_Enthalpy_Of_Formation.
- http://www.chemistry-reference.com/Standard%20Thermodynamic%20Values.pdf.
- J.-W. Zhao, H. Ding, X.-F. Tian, W.-J. Zhao and H.-L. Hou, Thermodynamic calculation on the formation of titanium hydride, Chin. J. Chem. Phys., 2008, 21, 569–574 CrossRef CAS.
- M. Arita, K. Shimizu and Y. Ichinose, Thermodynamics of the Ti–H system, Metall. Trans. A, 1982, 13, 1329–1336 CrossRef CAS.
- V. D. Anna, A. Spyratou, M. Sharma and H. Hagemann, FT-IR spectra of inorganic borohydrides, Spectrochim. Acta, Part A, 2014, 128, 902–906 CrossRef PubMed.
- T. D. Humphries, M. B. Ley, C. Frommen, K. T. Munroe, T. R. Jensen and B. C. Hauback, Crystal structure and in situ decomposition of Eu(BH4)2 and Sm(BH4)2, J. Mater. Chem. A, 2015, 3, 691–698 CAS , ESI.†.
- T. Sun, H. Wang, Q. Zhang, D. Sun, X. Yao and M. Zhu, Synergetic effects of hydrogenated Mg3La and TiCl3 on the dehydrogenation of LiBH4, J. Mater. Chem., 2011, 21, 9179–9184 RSC.
- M. Au, A. Jurgensen and K. Zeigler, Modified lithium borohydride for reversible hydrogen storage (2), J. Phys. Chem. B, 2006, 110, 26482–26487 CrossRef CAS PubMed.
- D. Liu, J. Yang, J. Ni and A. Drews, Studies of the effects of TiCl3 in LiBH4/CaH2/TiCl3 reversible hydrogen storage system, J. Alloys Compd., 2012, 514, 103–108 CrossRef CAS.
- K. Kadoi, N. Babcsań and H. Nakae, Heat treatment of TiH2 powder to control decomposition phenomenon for aluminum foam fabrication by melt route, Mater. Trans., 2009, 50, 727–733 CrossRef CAS.
- http://www.hydrogenfuelnews.com/new-hydrogen-fuel-cell-battery-could-help-power-mobile-devices/8523549/.
- http://www.ardica.com.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21539d |
| This journal is © The Royal Society of Chemistry 2016 |