
Prussian blue as positive electrode material for aqueous sodium-ion capacitor with excellent performance†
Lei Zhoua,
Zhengkai Yanga,
Chunyang Lia,
Bingwei Chenb,
Yanfeng Wangb,
Lijun Fua,
Yusong Zhu*a,
Xiang Liu*a and
Yuping Wu*ab
aCollege of Energy and Institute of Advanced Materials, Nanjing Tech University, Nanjing 211816, Jiangsu Province, China. E-mail: iamxliu@njtech.edu.cn
bNew Energy and Materials Laboratory, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: wuyp@fudan.edu.cn
First published on 24th October 2016
Abstract
High-quality Prussian blue (PB), whose crystals have a small number of vacancies, is synthesized by utilizing Na4Fe(CN)6 as the only precursor. It is demonstrated that the crystals have an important influence on electrochemical performance. The PB electrode exhibits a specific capacity as high as 107 mA h g−1 at a current density of 0.5 A g−1 without apparent capacity loss after 1100 cycles in 0.5 mol L−1 Na2SO4 aqueous solution due to fewer vacancies. Furthermore, a sodium-ion capacitor is also fabricated by combining the PB as a positive electrode and activated carbon as a negative electrode. It can operate at a cell voltage as high as 1.8 V with an energy density of 30 W h kg−1. The capacitor displays not only high energy density but also excellent cycling stability because the as-prepared PB has outstanding ion storage capability and excellent structural stability. These results show that the PB is a promising candidate for aqueous sodium-ion capacitors.
Introduction
Renewable energy sources, for example wind and solar power, have attracted much attention due to the concern of environmental protection and continuous exhaustion of fossil fuels.1,2 However, they are intermittent in nature while the demand of electric energy is possible at any time.3 Thus, the efficient utilization of these intermittent energy sources is dependent on smart grids and energy storage devices.4,5 Due to some advantages of power delivery and cycling life, electrochemical capacitors are becoming more and more indispensable as energy storage devices for electrical equipment, for instance cranes, forklifts, and electric vehicles.6–9 However, capacitors have an unsatisfactory low energy density. Metal-ion (such as Li-ion, Na-ion, Zn-ion, Mg-ion and Al-ion) capacitors have attracted attention on account of a much higher energy density ever since the first introduction of Li-ion capacitor in 2001.10 The ion-intercalation electrode offers larger energy density when the capacitive electrode shows excellent cycling stability.11 Although some attempts have been devoted to enhancing their energy density, most metal-ion capacitors present poor cycling stability and low power densities. Consequently, it remains a major challenge to develop energy storage systems with higher energy density and better cycling stability.Recently, Gogotsi's group and our lab found that both Ti3C2 and exfoliated graphite presented reversible cation (Na+, Li+, and Al3+) storage behavior in aqueous electrolytes.12–14 The research provides inspiration for developing intercalation electrodes for metal-ion capacitors. Meanwhile, sodium has more abundant reserves and has similar properties to lithium. Therefore, sodium is more suitable for large-scale applications in energy storage. In addition, an aqueous metal-ion capacitor is not only safe but also has low cost, and has potential for remarkable cycling stability.15–17 Some scientific and technical work has tried to replace lithium with sodium so as to build a Na-ion capacitor in recent years.18–22 For example, our group reported a quasi-solid-state sodium-ion capacitor (amorphous carbon/amorphous graphene) of excellent cycling stability with a 85% capacity retention over 1200 cycles in a gel polymer electrolyte containing 1 mol L−1 NaClO4/PC, and its energy density based on the weights of the active mass of two electrodes is 168 W h kg−1, which is comparable with those of Na-ion batteries.13 However, its cost is not low. Thus, a rational solution is to replace the organic electrolytes with aqueous ones. At present, there are only a few reports on metal-ion capacitors with Prussian blue (PB) analogues such as Li, Mg and Al.23–25
The general formula of PB is NaxFe[Fe(CN)6]y·□1−y·mH2O (0 < x < 2, y < 1). In the formula, the square □ expresses a [Fe(CN)6] vacancy taken up by coordinating water.26 The PB undergoes a two-electron redox reaction as follows:
| NaxFeIIIFeIII(CN)6 ↔ Na+ + Nax−1FeIIIFeII(CN)6 + e− ↔ 2Na+ + 2e− + Nax−2FeIIFeII(CN)6 | (1) |
These redox reactions could achieve high ion storage. The specific capacity of the PB Na2FeFe(CN)6 reaches 170 mA h g−1 in theory and the PB is suitable for large-scale applications by utilizing low-cost Na4Fe(CN)6 as the precursor.26–30 Furthermore, the open framework has large octahedral interstitial sites and ionic diffusion channels, which allow Na-ion insertion/extraction with integrality of the crystal framework.31–35 Therefore, PB is a promising material for the positive electrode of Na-ion capacitors. In addition, [Fe(CN)6]4− vacancies taken up by coordinated water in PB may give rise to lattice distortion and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bridge collapse during the Na-ion insertion/extraction process.36 Consequently, it is very necessary to prepare high-quality PB crystals with few vacancies. Recently, Guo reported that high-quality Na0.61Fe[Fe(CN)6]0.94 PB crystals could be synthesized by a simple method using Na4Fe(CN)6 as the only iron source and could be cycled over 150 cycles without decay in 1 mol L−1 NaPF6/PC/DEC electrolyte.37 At the same time, Dou's group reported a Na-enriched framework, Na1.56Fe[Fe(CN)6], with few vacancies, which was synthesized by a facile one-step method, and it could be cycled up to 400 cycles without capacity loss in an organic electrolyte.38 These reports mainly introduced the electrochemical performances of NaxFeIIIFeII(CN)6 for sodium-ion batteries in organic electrolytes. However, it is still unknown what performance NaxFeIIIFeII(CN)6 would display in aqueous electrolytes.
N bridge collapse during the Na-ion insertion/extraction process.36 Consequently, it is very necessary to prepare high-quality PB crystals with few vacancies. Recently, Guo reported that high-quality Na0.61Fe[Fe(CN)6]0.94 PB crystals could be synthesized by a simple method using Na4Fe(CN)6 as the only iron source and could be cycled over 150 cycles without decay in 1 mol L−1 NaPF6/PC/DEC electrolyte.37 At the same time, Dou's group reported a Na-enriched framework, Na1.56Fe[Fe(CN)6], with few vacancies, which was synthesized by a facile one-step method, and it could be cycled up to 400 cycles without capacity loss in an organic electrolyte.38 These reports mainly introduced the electrochemical performances of NaxFeIIIFeII(CN)6 for sodium-ion batteries in organic electrolytes. However, it is still unknown what performance NaxFeIIIFeII(CN)6 would display in aqueous electrolytes.
In this paper, high-quality PB was synthesized by a facile one-step method utilizing Na4Fe(CN)6 as the only iron source. In this strategy, the synthesized PB presented only a small number of vacancies in its crystal structure due to the slow growth process, which has a positive effect on ion-storage capability and the stability of crystal framework. It turned out that fewer vacancies could not only improve its cycling stability but also enhance its specific capacity in an aqueous electrolyte.
Experimental section
Preparation of Prussian blue
PB crystals were synthesized by a facile one-step method, as illustrated in Fig. S1 (ESI†). All chemicals were bought from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China) without further treatment. In a typical synthesis, 1 g NaCl was dissolved into 100 mL deionized water. Then, 2 mL HCl (37%) was added into sodium chloride (NaCl) solution. Later, 0.580 g Na4Fe(CN)6·10H2O and 1 g poly(vinyl pyrrolidone) were added into the mixed solution under ultrasonic stirring. After vigorous stirring for 1 h, a clear blue solution was obtained. The reaction was carried out for 10 h at 80 °C. The sample was collected by centrifugation at 8000 rpm for 10 min and washed in distilled water and ethanol three times. After the sample was dried at 100 °C in a vacuum oven for 24 h, PB cubes were obtained.Characterization of Prussian blue
X-ray diffraction (XRD) measurements were carried out by using a Bruker D4 X-ray diffractometer (Bruker, Germany) with Ni-filtered Cu-Kα radiation (40 kV, 40 mA). Scanning electron microscopy images were obtained by a Philips XL30 microscope (Philips, Netherlands) operated at 25 kV. Inductively coupled plasma (ICP) analysis was conducted by a Thermo E.IRIS Duo. The elemental analysis for C and N elements was conducted with a Vario EL elemental analyzer.Electrochemical measurements
The synthesized PB was mixed with acetylene black (conductive agent) and polytetrafluoroethylene (binder) in a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 with ethanol. After drying, the mixtures were pressed into films, and then the films were cut into round disks and dried at 80 °C for 10 h. These disks were fixed onto graphite felt to act as working electrodes and the active mass loading was about 1.5 mg. Cyclic voltammetry (CV) and charge–discharge measurements of the PB electrodes were performed in 0.5 M Na2SO4 aqueous solution with three-electrode cells, in which graphite felt and saturated calomel electrode (SCE) were used as the counter and the reference electrodes, respectively. The CV curves were collected with an electrochemical workstation (CHI660C, Chenhua, China). An activated carbon (AC) with a specific surface area of about 2800 m2 g−1 was purchased from Ningde Xinseng Chemical Industrial Co. Ltd (China). The AC electrode was prepared in the same way as the above PB electrode. A two-electrode cell, which consisted of the PB positive electrode and the AC negative electrode in 0.5 M Na2SO4 aqueous solution, was used to test the cycling behavior with a Land cell tester (Wuhan, China).
:10:10 with ethanol. After drying, the mixtures were pressed into films, and then the films were cut into round disks and dried at 80 °C for 10 h. These disks were fixed onto graphite felt to act as working electrodes and the active mass loading was about 1.5 mg. Cyclic voltammetry (CV) and charge–discharge measurements of the PB electrodes were performed in 0.5 M Na2SO4 aqueous solution with three-electrode cells, in which graphite felt and saturated calomel electrode (SCE) were used as the counter and the reference electrodes, respectively. The CV curves were collected with an electrochemical workstation (CHI660C, Chenhua, China). An activated carbon (AC) with a specific surface area of about 2800 m2 g−1 was purchased from Ningde Xinseng Chemical Industrial Co. Ltd (China). The AC electrode was prepared in the same way as the above PB electrode. A two-electrode cell, which consisted of the PB positive electrode and the AC negative electrode in 0.5 M Na2SO4 aqueous solution, was used to test the cycling behavior with a Land cell tester (Wuhan, China).
Results and discussion
The XRD pattern of the prepared PB is shown in Fig. 1a. It is a face-centered cubic phase without secondary phases, and the characteristic peaks such as (200), (220), (400), (420), (422), (440), (600), and (620) can be clearly identified, indicating good crystal structure. The lattice parameter of the PB was refined to be a = 10.2 Å. PB cubes were formed and have a good size distribution of 2–3 μm as displayed in the micrograph of Fig. 1b. The chemical formula of PB sample was determined by ICP analysis for elements Fe and Na, and CHN elemental analysis for N and C. On the basis of contents of Fe, Na, N, and C for the sample (ESI, Table S1†), the chemical formula for the fabricated PB is Na1.29Fe[Fe(CN)6]0.91·□0.09 and it demonstrates that the [Fe(CN)6]4− vacancy content in PB is only 9%.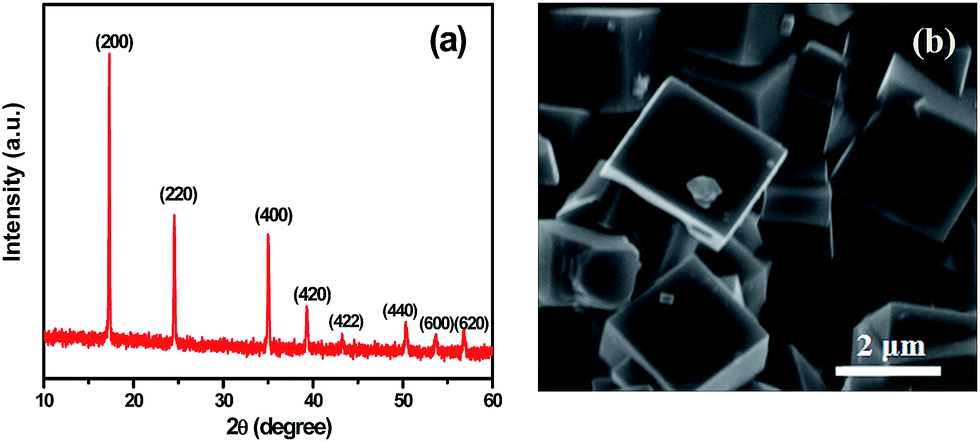 | ||
| Fig. 1 Some physical characteristics of the prepared PB: (a) XRD pattern and (b) scanning electron micrograph. | ||
The prepared PB has perfect cubic crystals with few vacancies. Some Na ions would have access to enter into the open framework of the PB during the preparation process in NaCl solution, which could reduce the vacancies as well as increase the Na ion amount in the PB. For one thing, the reduction of vacancies enhances cycling stability and also improves coulombic efficiency. For another thing, more Na ions entering into the open framework could also improve specific capacity. During the preparation process, Na4[Fe(CN)6] was gradually dissolved into Fe2+ in an acidic environment, and then Fe2+ was oxidized to Fe3+ step by step owing to its instability. Next, Fe2+ and Fe3+ combined with undecomposed [Fe(CN)6]4− to fabricate the nuclei of PB. Finally, the size of PB cubes gradually grew with the reaction time.
As shown in Fig. 2, the electrochemical performance of the PB was studied in the three-electrode system. The CV curve of the PB (Fig. 2a) shows two pairs of sharp and symmetric oxidation–reduction peaks, which are situated at the high potential region of 0.9–1.2 V (vs. SCE) and at the low potential region of −0.2–0.3 V (vs. SCE), respectively. The shapes and potential positions of these CV peaks remain almost unchanged at a scan rate of 5 mV s−1 during successive scans, which suggests stable and reversible Na-ion insertion/extraction reactions. In addition, the CV curves are not the same at different scan rate and reveal that the polarization or over-potential still exists (ESI, Fig. S2†).
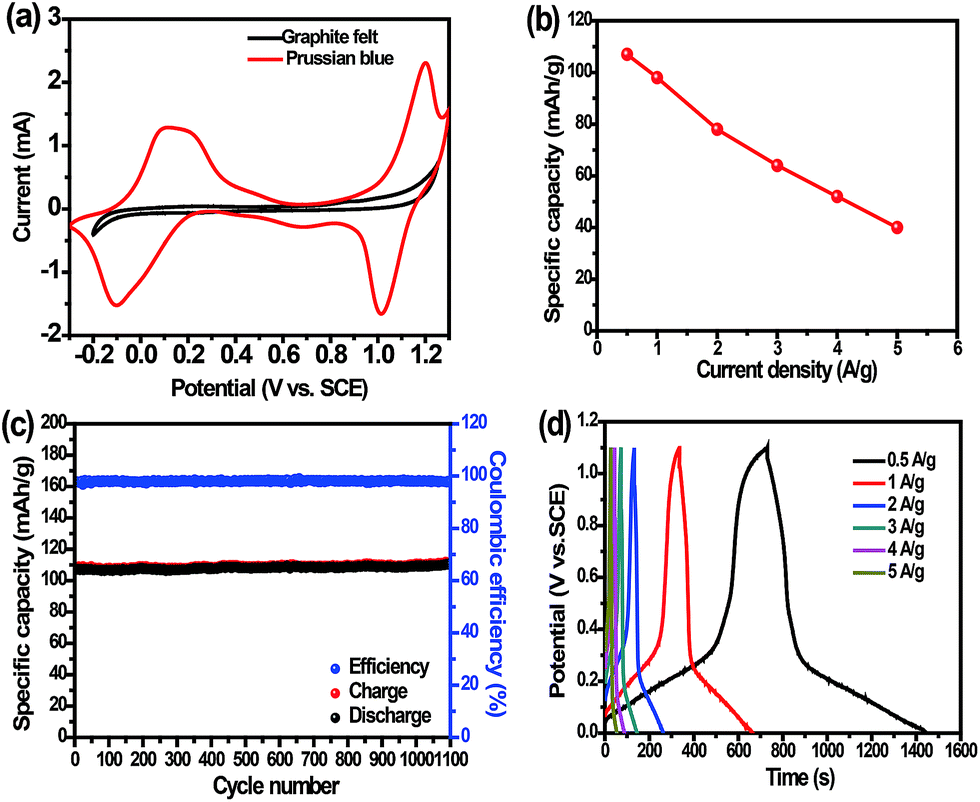 | ||
| Fig. 2 Electrochemical properties of the PB. (a) CV curves of PB and graphite felt, (b) specific capacity of the PB at different current densities, (c) cycling behavior at a current density of 0.5 A g−1, and (d) galvanostatic charge/discharge curves in the potential range from 0 to 1.1 V at different current densities. | ||
In order to further explore whether the insertion and extraction of Na ions truly occurs in PB, the PB samples at different charge and discharge states were characterized by XRD as shown in Fig. S4 (ESI†). On the one hand, the XRD peaks shift gradually toward higher position (decrease of the lattice parameter) during the charge process. On the other hand, the XRD peaks move gradually toward smaller angle (increase of the lattice parameter) during the discharge process. As can be seen from Fig. 3a and b, the (200) and (220) peaks in each sample uniformly shift to higher angles with increasing Na-ion concentration during Na-ion insertion processes, and to lower angles with decreasing Na-ion concentration during Na-ion extraction processes. This phenomenon demonstrates clearly that the crystal structure of PB generates phase transitions from cubic to rhombohedral,37 leading to a decrease of its symmetry and a consequent decrease of lattice parameters. This is similar to spinel LiMn2O4 for positive electrodes of lithium ion batteries.39 Meanwhile, the change of crystal structure is highly reversible during the Na-ion insertion/extraction process. A schematic illustration of the PB redox mechanism is shown in Fig. 4. The PB goes through a two-electron redox reaction as follows:
| FeIIIFeIII ↔ FeIIIFeII ↔ FeIIFeII | (2) |
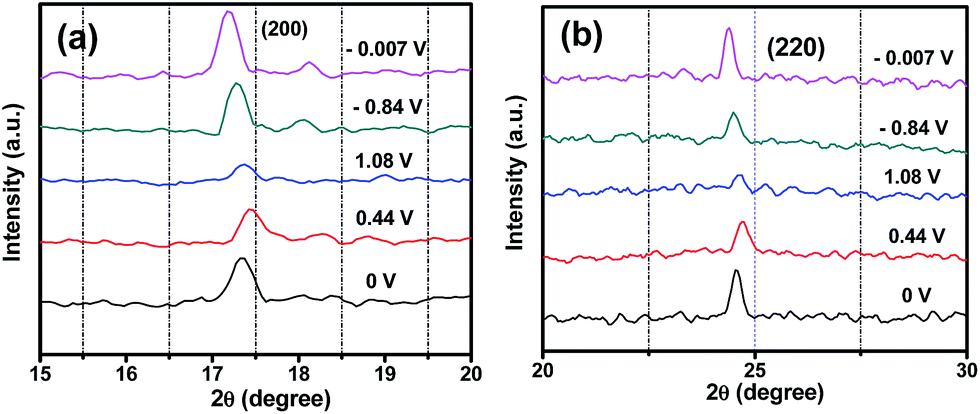 | ||
| Fig. 3 XRD patterns of the PB electrode at various charge and discharge states (a) in the range of 15–20° and (b) in the range of 20–30°. | ||
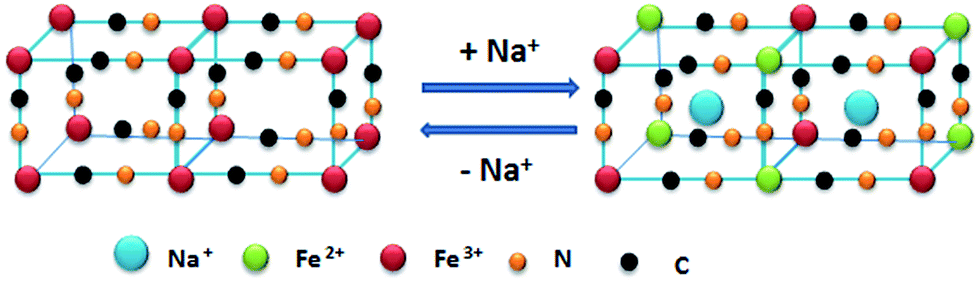 | ||
| Fig. 4 Schematic illustration of the redox mechanism of PB from reversible sodium-ion insertion and extraction in Na2SO4 electrolyte solution. | ||
The perfect structure of PB with good reversibility and stability is favored. The reason is that the redox reaction promotes the resistance to mechanical strain and volume variation and prevents the collapse of the crystal framework during sodium-ion insertion/extraction process.37
Fig. 2b shows the specific capacity of the PB electrode at different current densities. When the current density increases from 0.5 to 5 A g−1, the reversible capacity reduces gradually from 107 to 40 mA h g−1. Even at a high current density of 5 A g−1, the capacity retention is still up to 37%. It is apparent that electrochemical performance of the as-prepared PB is similar to that of a PB analogue (CuHCF) sample reported by Cui's group,40 but ours presents higher reversible capacity. Fig. 2c presents superior cycling stability, and there is no apparent capacity loss after 1100 cycles at a current density of 0.5 A g−1. It can be seen that the discharge capacity can reach 107 mA h g−1 between 0 to 1.1 V (vs. SCE) and the coulombic efficiency remains at almost 100% at a current density of 0.5 A g−1 even in the first cycle (ESI, Fig. S3a†). Furthermore, our prepared PB electrode can give ultrahigh charge specific capacity (160 mA h g−1) between 0 and 1.2 V (vs. SCE) without regard to coulombic efficiency in an aqueous electrolyte, which approaches its theoretical specific capacity (ESI, Fig. S3b†). Although the discharge capacity of the PB is lower than those of PB reported in organic electrolytes,41–48 the reversible capacity is higher than those of PB analogues reported in aqueous electrolytes so far. Generally speaking, the capacity of an electrode in an organic electrolyte is higher than the same electrode in aqueous solution.39 For instance, a special nanotube LiMn2O4 electrode gives a higher capacity of 110 mA h g−1 at a current density of 0.5 A g−1 in aqueous solution, but a general LiMn2O4 electrode can exhibit a capacity as high as 120–140 mA h g−1 at 1C rate in organic electrolyte.49,50 The high-enough capacity is derived from effective utilization of the two-electron redox reactions of the Fe2+/Fe3+ couple and vacancy-free lattice as displayed in Fig. 4. The outstanding cyclability arises from the channeled and stable framework of the PB, which offers very many sites for reversible Na-ion insertion/extraction without obvious structural fading. Fig. 2d exhibits galvanostatic charge/discharge curves in the potential range from 0 to 1.1 V at different current densities. A distinct discharging platform can be found at the lower potential region of 0–0.2 V, which is consistent with the CV curves. Due to the narrow charge/discharge potential region and polarization, the platform at higher potential is not obvious at these current densities.
As shown in Fig. 5, the Nyquist plot of the PB electrode in the frequency range from 105 to 10−2 Hz displays a semicircular arc at mid-high frequency and a straight line at low frequency. Generally speaking, an angle of about 45° in the linear region is related to the diffusion process of ions in the electrode. However, the linear region of the PB electrode shows an angle of about 60° corresponding to the real axis, demonstrating that the electrode process is not completely under diffusion control and it also exhibits capacitive behavior.51 The X-intercept of the Nyquist plot is related to solution resistance (Rs), which contains ionic and electronic resistances,52 and the semicircular arc is the result of charge-transfer resistance (Rct) and double-layer capacitance.53 The Rct can be calculated from the diameter of the semicircle on the axes. The measured values of Rs and Rct for PB are 7.5 and 39 Ω cm−2, respectively. It can be seen that PB exhibits a small Rct, which is one reason why the PB presents a faster charge–discharge behavior.
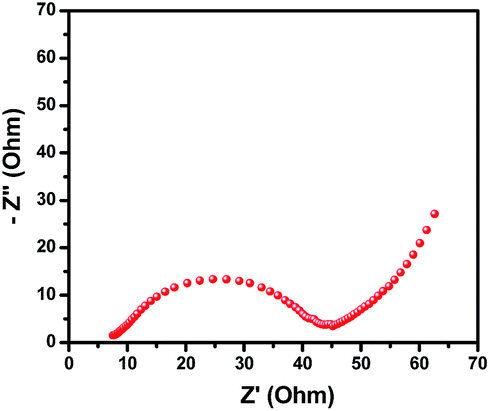 | ||
| Fig. 5 Nyquist plot of PB in 0.5 M Na2SO4 aqueous electrolyte in the frequency range from 105 to 10−2 Hz. | ||
An aqueous Na-ion capacitor is assembled by using the PB positive electrode combined with an AC negative electrode with a mass ratio of 1:3. As exhibited in Fig. 6, the electrochemical performance of this capacitor was investigated in a two-electrode system. As shown in Fig. 6a, the CV curve presents two pairs of peaks at high potential voltage (0–0.5 V) and at low potential voltage (1.5–1.9 V). The sodium-ion capacitor delivers a specific capacity of 100 mA h g−1 (based on the weight of the PB positive electrode) at 0.5 A g−1 (see Fig. 6b), which is lower than that of the single PB electrode. As seen from Fig. 6c, the long-term cyclability of this capacitor is measured in the charge/discharge voltage from 0 to 1.8 V at a current density of 0.5 A g−1, which keeps stable after 1000 cycles with a high capacity retention of 97%. Moreover, Fig. S5 (ESI†) exhibits electrochemical impedance spectra of the PB/AC capacitor before and after 1000 cycles. The semicircular diameter became larger after the cycling test, which demonstrates that Na-ion insertion/extraction has become difficult in the positive electrode.54 The slope of the straight line is close to 90°, which indicates that a capacitive behavior occurs on the electrode surface associated with Na-ion adsorption/desorption.54,55 Next, we performed galvanostatic charge/discharge tests at current densities ranging from 0.5 A g−1 to 5 A g−1 (Fig. 6d). The charge/discharge curves reveal a smaller voltage drop at higher discharging current (5 A g−1) due to the smaller internal resistance of the capacitor, as exhibited in the inset to Fig. 6d.
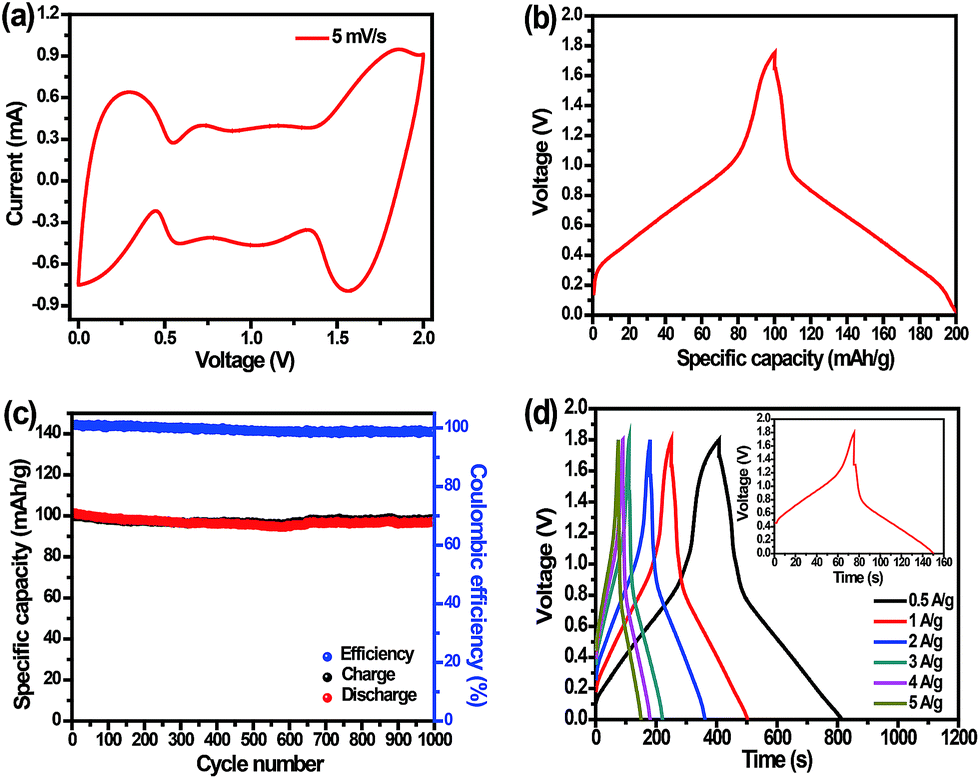 | ||
| Fig. 6 Electrochemical performance of the assembled Na-ion capacitor. (a) CV curve at a scan rate of 5 mV s−1, (b) typical galvanostatic charge/discharge curve at a current density of 0.5 A g−1, (c) the cycling behavior at the current density of 0.5 A g−1, and (d) galvanostatic charge/discharge curves in the voltage range from 0 to 1.8 V at different current densities. Inset: galvanostatic charge/discharge curve at a current density of 5 A g−1. | ||
Fig. 7a presents the working mechanism of the sodium-ion capacitor. This energy storage system couples a high capacity intercalation compound-based battery-type positive electrode (PB) and a high rate negative electrode (AC) based on capacitor-type surface adsorption. Fig. 7b exhibits the specific capacity of the aqueous Na-ion capacitor at different current density (from 0.5 to 5 A g−1). The capacitor can deliver a reversible capacity of 100, 94, 79, 56, and 46 mA h g−1 on the basis of the positive electrode at 0.5, 1, 2, 3, and 4 A g−1, respectively. Even at the higher current density of 5 A g−1, the reversible capacity still reaches 33 mA h g−1, corresponding to 35% capacity utilization. Fig. 7b also reveals the relation between the power density and the energy density of the aqueous sodium-ion capacitor, which are calculated based on the total weight of active materials. The energy density of the capacitor is 30 W h kg−1 at a power density of 423 W kg−1, and it can still maintain a value of 10 W h kg−1 even at a higher power of 1193 W kg−1. In some cases, the reported energy density is only based on the positive electrode. If it is applied in our case, the energy density will reach up to 90 W h kg−1 only based on the positive electrode, which is superior to the reported data for some PB analogs. The higher energy density with long cycling life is superior to conventional aqueous rechargeable capacitors. Consequently, the material enables the sodium-ion capacitor to serve as a clean and low-cost alternative for large-scale electricity storage.
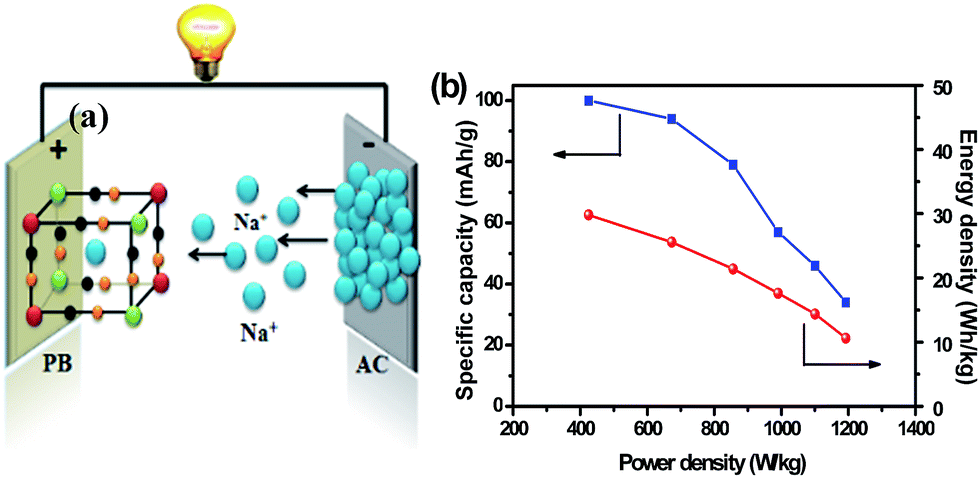 | ||
| Fig. 7 (a) Schematic of the working mechanism of a sodium-ion capacitor, and (b) Ragone plots of the aqueous sodium-ion capacitor in 0.5 M Na2SO4 electrolyte. | ||
Conclusions
In summary, we have successfully fabricated high-quality cubic PB crystals with few vacancies via a simple and cost-effective strategy. The unique crystal architecture plays a crucial role in active sites for faradaic reactions. On the one hand, the ordered lattice provides a large number of interstitial active sites for sodium-ion insertion during cycling, which results in higher reversible capacity and coulombic efficiency. On the other hand, it can also prevent framework collapse and provide excellent cycling performance in aqueous solution. Thus, the PB electrode gives a prominent specific capacity as high as 107 mA h g−1 for sodium ions, much higher than those of other PB derivatives for sodium ions in aqueous solution. And, there is no apparent capacity loss after 1100 cycles at a current density of 0.5 A g−1. Furthermore, we also assembled a sodium-ion capacitor, which consisted of AC as negative electrode together with PB positive electrode in 0.5 mol L−1 Na2SO4 aqueous electrolyte. The sodium-ion capacitor shows a high voltage of 1.8 V and an energy density of 30 W h kg−1 on the basis of the total active electrode weights. Moreover, the sodium-ion capacitor shows excellent rate performance and good cycling stability. Last but not least, both PB and AC are of low cost and environmentally friendly, making the Na-ion capacitor promising for power storage applications.Acknowledgements
This work is supported by the Distinguished Young Scientists Program of the National Natural Science Foundation of China (NSFC51425301).Notes and references
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed
.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed
- X. N. Xie, Y. Wang, Q. Wang and K. P. Loh, Adv. Mater., 2012, 24, 76–81 CrossRef CAS PubMed
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC
- Y. M. Chen, Z. Li and X. W. D. Lou, Angew. Chem., Int. Ed., 2015, 127, 10667–10670 CrossRef
- G. G. Amatucci, F. Badway, A. D. Pasquier, T. Zheng and J. Dahn, J. Electrochem. Soc., 2001, 148, A930–A939 CrossRef CAS
- V. Aravindan, J. Gnanaraj, Y. S. Lee and S. Madhavi, Chem. Rev., 2014, 114, 11619–11635 CrossRef CAS PubMed
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. D. Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed
- F. X. Wang, X. W. Wang, Z. Chang, X. Wu, X. Liu, L. J. Fu, Y. S. Zhu, Y. P. Wu and W. Huang, Adv. Mater., 2015, 27, 6962–6968 CrossRef CAS PubMed
- F. X. Wang, Z. C. Liu, X. W. Wang, X. H. Yuan, X. W. Wu, Y. S. Zhu, L. J. Fu and Y. P. Wu, J. Mater. Chem. A, 2016, 4, 5115–5123 CAS
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed
- F. X. Wang, S. Y. Xiao, Y. S. Zhu, Z. Chang, C. L. Hu, Y. P. Wu and R. Holze, J. Power Sources, 2014, 246, 19–23 CrossRef CAS
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS
- V. Aravindan, M. Ulaganathan and S. Madhavi, J. Mater. Chem. A, 2016, 4, 7538–7548 CAS
- J. Ding, H. Wang, Z. Li, K. Cui, D. Karpuzov, X. Tan, A. Kohandehghan and D. Mitlin, Energy Environ. Sci., 2015, 8, 94–955 Search PubMed
- Z. Chen, V. Augustyn, X. Jia, Q. Xiao, B. Dunn and Y. Lu, ACS Nano, 2012, 6, 4319–4327 CrossRef CAS PubMed
- S. Banerjee, S. Neihsial and S. K. Pati, J. Mater. Chem. A, 2016, 4, 5517–5527 CAS
- V. Aravindan, M. Ulaganathan and S. Madhavi, J. Mater. Chem. A, 2016, 4, 7538–7548 CAS
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, H. S. Zhou and K. Oh-ishi, J. Phys. Chem. C, 2013, 117, 10877–10882 CAS
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, K. Oh-ishi, A. Okazawa, N. Kojima, R. Kurono, S. Nishimurac and A. Yamada, J. Mater. Chem. A, 2013, 1, 13055–13059 CAS
- Z. Li, K. Xiang, W. T. Xing, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2015, 5, 1401410 CrossRef
- D. Asakura, M. Okubo, Y. Mizuno, T. Kudo, H. Zhou, K. Ikedo, T. Mizokawa, A. Okazawa and N. Kojima, J. Phys. Chem. C, 2012, 116, 8364–8369 CAS
- M. Okubo, D. Asakura, Y. Mizuno, J. D. Kim, T. Mizokawa, T. Kudo and I. Honma, J. Phys. Chem. Lett., 2010, 1, 2063–2071 CrossRef CAS
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef PubMed
- Y. Mizuno, M. Okubo, K. Kagesawa, D. Asakura, T. Kudo, H. Zhou, K. Oh-ishi, A. Okazawa and N. Kojima, Inorg. Chem., 2012, 51, 10311–10316 CrossRef CAS PubMed
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, H. Zhou and K. Ohishi, J. Phys. Chem. C, 2013, 117, 10877–10882 CAS
- T. Matsuda, M. Takachi and Y. Moritomo, Chem. Commun., 2013, 49, 2750–2752 RSC
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, Angew. Chem., Int. Ed., 2013, 52, 1964–1967 CrossRef CAS PubMed
- M. Zhou, J. Qian, X. Ai and H. Yang, Adv. Mater., 2011, 23, 4913–4917 CrossRef CAS PubMed
- H. Lee, Y. I. Kim, J. K. Park and J. W. Choi, Chem. Commun., 2012, 48, 8416 RSC
- D. Asakura, M. Okubo, Y. Mizuno, T. Kudo, H. Zhou, K. Ikedo, T. Mizokawa, A. Okazawa and N. Kojima, J. Phys. Chem. C, 2012, 116, 8364–8369 CAS
- X. Wu, W. Deng, J. Qian, Y. Cao, X. Ai and H. Yang, J. Mater. Chem. A, 2013, 1, 10130–10134 CAS
- Y. You, X. L. Wu, Y. X. Yin and Y. G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 CAS
- W. J. Li, S. L. Chou, J. Z. Wang, Y. M. Kang, J. L. Wang, Y. Liu, Q. F. Gu, H. K. Liu and S. X. Dou, Chem. Mater., 2015, 27, 1997–2003 CrossRef CAS
- Y. P. Wu, Lithium-Ion Batteries: Fundamentals and Applications, CRC Press, New York, 2015 Search PubMed
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef PubMed
- X. Y. Wu, M. Y. Sun, Y. F. Shen, J. F. Qian, Y. L Cao, X. P. Ai and H. X. Yang, ChemSusChem, 2014, 7, 407–411 CrossRef CAS PubMed
- D. J. Kim, Y. H. Jung, K. K. Bharathi, S. H. Je, D. K. Kim, A. Coskun and J. W. Choi, Adv. Energy Mater., 2014, 4, 1400133 CrossRef
- Y. F. Yue, A. J. Binder, B. K. Guo, Z. Y. Zhang, Z. A. Qiao, C. C. Tian and S. Dai, Angew. Chem., Int. Ed., 2014, 53, 3134–3137 CrossRef CAS PubMed
- Y. H. Lu, L. Wang, J. G. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC
- M. Okubo, C. H. Li and D. R. Talham, Chem. Commun., 2014, 50, 1353–1355 RSC
- D. Yang, J. Xu, X. Z. Liao, Y. S. He, H. Liu and Z. F. Ma, Chem. Commun., 2014, 50, 13377–13380 RSC
- L. Wang, J. Song, R. Qiao, L. A. Wray, M. A. Hossain, Y. D. Chung, W. Yang, Y. Lu, D. Evans, J. J. Lee, S. Vail, X. Zhao, M. Nishijima, S. Kakimoto and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2548–2554 CrossRef CAS PubMed
- S. Yu, Y. Li, Y. Lu, B. Xu, Q. Wang, M. Yan and Y. Jing, J. Power Sources, 2015, 275, 45–49 CrossRef CAS
- W. Tang, Y. Y. Hou, F. X. Wang, L. L. Liu, Y. P. Wu and K. Zhu, Nano Lett., 2013, 13, 2036–2040 CrossRef CAS PubMed
- W. W. Sun, H. Q. Liu, T. Peng, Y. M. Liu, G. X. Bai, S. Kong, S. S. Guo, M. Y. Li and X. Z. Zhao, J. Mater. Chem. A, 2015, 3, 8165–8170 CAS
- Q. T. Qu, P. Zhang, B. Wang, Y. Chen, S. Tian, Y. P. Wu and R. Holze, J. Phys. Chem. C, 2009, 113, 14020–14027 CAS
- G. S. Gund, D. P. Dubal, S. S. Shinde and C. D. Lokhande, ACS Appl. Mater. Interfaces, 2014, 6, 3176–3188 CAS
- Q. T. Qu, L. J. Fu, X. Y. Zhan, D. Samuelis, J. Maier, L. Li, S. Tian, Z. H. Li and Y. P. Wu, Energy Environ. Sci., 2011, 4, 3985–3990 CAS
- Y. Zhang, C. Yuan, K. Ye, X. Jiang, J. Yin, G. Wang and D. Cao, Electrochim. Acta, 2014, 148, 237–243 CrossRef CAS
- Y. Zhang, K. Ye, K. Cheng, G. Wang and D. Cao, Electrochim. Acta, 2014, 148, 195–202 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21500a |
| This journal is © The Royal Society of Chemistry 2016 |