
Photophysical properties of asymmetric and water-soluble dinuclear lanthanide complexes of poly glycol chain functionalized-benzoic acid derivative: experimental and theoretical approaches†
Abstract
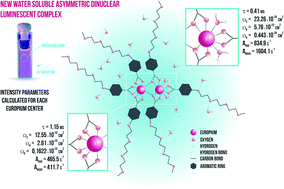
New water-soluble lanthanide complexes with a poly glycol chain (–OEtOEtOEtOMe) functionalized benzoic acid derivative ligand were synthesized and photophysically characterized. The results indicate a formation of an asymmetric dinuclear lanthanide complexes. The [Eu2(mee)6(H2O)2] complex shows emission in water solution and it was noted that after 48 h part of the ligands are replaced by water molecules resulting on quenching of the emission and decreasing of lifetimes by O–H oscillators from water molecules. The average Judd–Ofelt intensity parameters were determined experimentally from emission spectra of the europium(III) complex. Additionally, the contribution of each asymmetric europium(III) center to the JO intensity parameters were calculated independently using time resolved spectroscopy and the LUMPAC software helping the proposition of the polyhedral coordination sphere determined by the ground state geometry using the Sparkle/PM3 model. The transfer and back energy transfer rates were also calculated. The values obtained for energy transfer rates are lower than previous values obtained for europium(III) complexes with benzoic acid derivative ligands, indicating the role of the poly glycol chain on photophysical properties of the [Eu2(mee)6(H2O)2] complex.
 Please wait while we load your content...
Please wait while we load your content...

