
2-Aminoimidazole borohydride as a hydrogen carrier†
Yong Wu,
Yue Qi,
Jun Chen,
He Fu,
Jie Zheng* and
Xingguo Li*
Beijing National Laboratory for Molecular Science (BNLMS), College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: xgli@pku.edu.cn; Fax: +86-10-62765930; Tel: +86-10-62765930
First published on 24th October 2016
Abstract
2-Aminoimidazole borohydride (Im-NH2BH4) is synthesized via the reaction between 2-aminoimidazole hemisulfate ((Im-NH2)2SO4) and sodium borohydride by a simple ball milling method. This compound holds theoretical hydrogen capacity of 8.1 wt%. It is designed based on the strategy of destabilizating BH4− using a large conjugated cation, Im-NH2+ and maximizing the protonic-hydridic hydrogen interaction by balancing their numbers. Thermal dehydrogenation analyses demonstrate that it can release about 3.2 wt% hydrogen (Na2SO4 included) without gaseous impurities below 320 °C. The decomposition process has three exothermal steps and the onset dehydrogenation temperature is 50.5 °C. These results indicate that the above strategy is effective. The dehydrogenation process of this compound is discussed and the product at 320 °C is proposed to be C3N3H3BH.
Introduction
Hydrogen is regarded as one of the most promising sources of energy for the future. However, effective storage of hydrogen is a giant obstacle for its wide application.1–5 In the last two decades, a large number of hydrogen storage materials, such as metal hydrides,6–11 complex hydrides,12–15 chemical hydrides,16,17 carbon materials,18–20 metal–organic frameworks,21,22 liquid organic hydrogen carriers,23,24 Kubas-type materials25,26 and their combinations, have been researched. Unfortunately, none of them can fulfil all the targets of the US Department of Energy (DOE).Metal borohydrides are promising hydrogen storage materials owing to their very high gravimetric hydrogen capacities. However, they suffer from high dehydrogenation temperature and release of borane.27 Many strategies are used to solve these issues, such as using additives,28–30 borohydrides with mixed cations31,32 and metal borohydride organic amine complexes.33–35 Some of the above attempts have encouraging results.
Recently, we reported that the dehydrogenation temperature of LiBH4 and NaBH4 can be dramatically reduced by dissolving them in a imidazolium-type ionic liquids.36 This originates from the destabilization of BH4− due to the more favourable charge transfer effect between BH4− and 1-n-butyl-3-methylimidazolium (bmim+), a large conjugated cation. However, the poor solubility of the metal borohydrides in the ionic liquid results in very low total hydrogen capacity. Nevertheless, it provides a new strategy to lower the dehydrogenation temperature of borohydrides.
Another strategy to promote the dehydrogenation of borohydrides is to utilize the protonic-hydridic hydrogen interaction. The borohydride–amine composites are largely based on this strategy. Moreover, guanidinium (Gua+, C(NH2)3+) type of borohydrides, such as methylguanidinium borodride,37 guanidinium borodride (GuaBH4),38,39 guanidinium octahydrotriborate (GuaB3H8),40 have been studied as hydrogen storage materials. Their dehydrogenation temperatures are also low mainly due to the interaction between the protonic H (H+) on N and the hydridic H (H−) on B atoms. However, dehydrogenation of GuaBH4 releases some NH3, due to the excess of the protonic H. This can be solved by adding proper amount of Ca(BH4)2 or LiBH4 to balance the number of H from N–H and B–H.41,42
Inspired by the above strategies, here we report 2-aminoimidazole borohydride (Im-NH2BH4, as shown in Fig. 1) as a novel hydrogen carrier. It consists of BH4− and a large conjugated cation, similar to the borohydrides dissolved in imidazolium-type ionic liquids. The amino group on the imidazole ring is to balance the number of protonic and hydridic hydrogen to maximize the H+–H− interaction. The theoretic hydrogen capacity of Im-NH2BH4 reaches 8.1 wt% and it can release up to 68% hydrogen below 320 °C, making it a promising hydrogen carrier.
 | ||
| Fig. 1 Schematic diagram of the structure of 2-aminoimidazole borohydride (yellow, B; grey, C; blue, N; white, H). | ||
Experimental section
Materials
(Im-NH2)2SO4 was purchased from Adamas Reagents Co. Ltd. and NaBH4 was purchased from Sigma-Aldrich Co. LLC. They were stored in an argon-filled glove box and used as-received without further purification.Synthesis of Im-NH2BH4
Im-NH2BH4 was synthesized by ball milling a mixture of (Im-NH2)2SO4 and NaBH4 in 1 bar argon atmosphere using a planetary ball milling apparatus (Pulverisette 5) for 40 min. The molar ratio of NaBH4 and (Im-NH2)2SO4 was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the mass ratio of balls to raw materials was 40:1. The effect of ball milling conditions was studied.
:1 and the mass ratio of balls to raw materials was 40:1. The effect of ball milling conditions was studied.
Characterization
The samples were characterized by powder X-ray diffraction (XRD, PANalytical X'Pert3 Powder, Cu Kα), Fourier transform infrared spectroscopy (FT-IR, Bruker Tensor 27 Fourier transform infrared spectrometer), solid-state nuclear magnetic resonance (SSNMR, AVANCE III 400 MHz WB solid-state NMR spectrometer, more details are available in the ESI†) and differential scanning calorimetry (DSC, NETZSCH DSC 204 HP calorimeter, heating rate of 5 K min−1, argon flow).The dehydrogenation process was characterized by temperature programmed desorption/mass spectrum (TPD/MS, Quantachrome Autosorb iQ automatic gas sorption analyser). The dehydrogenation kinetics was studied in a home-made Sievert type setup. The dehydrogenation capacity was quantified by a thermal conductivity detector (TCD) on the Quantachrome Autosorb iQ automatic gas sorption analyzer and also the Sievert's method as previously reported.36 The TCD signal is calibrated using hydrogen as the external standard (Fig. S1†).
Results and discussion
The synthesis and purification of Im-NH2BH4
Fig. 2 shows the XRD patterns of (Im-NH2)2SO4, NaBH4 and the samples after ball milling with different rotating speeds. Most raw materials remain unchanged when the rotating speed is 100 rpm and 150 rpm. In the case of 200 rpm, most of the starting materials are transformed and Na2SO4 is observed. The peaks are ascribed to two different phases of Na2SO4, phase V and phase III. The former is the thermodynamically stable phase at room temperature, while the latter is a metastable phase.43,44 The appearance of phase III may originate from the high energy process during the reaction under ball milling condition. There is some NaBH4 remaining, as indicated by the peak at 28.9°. However, no other new phases are observed, which indicates that the generated Im-NH2BH4 might be amorphous. The sample with 200 rpm is denoted as Im-NH2-RT.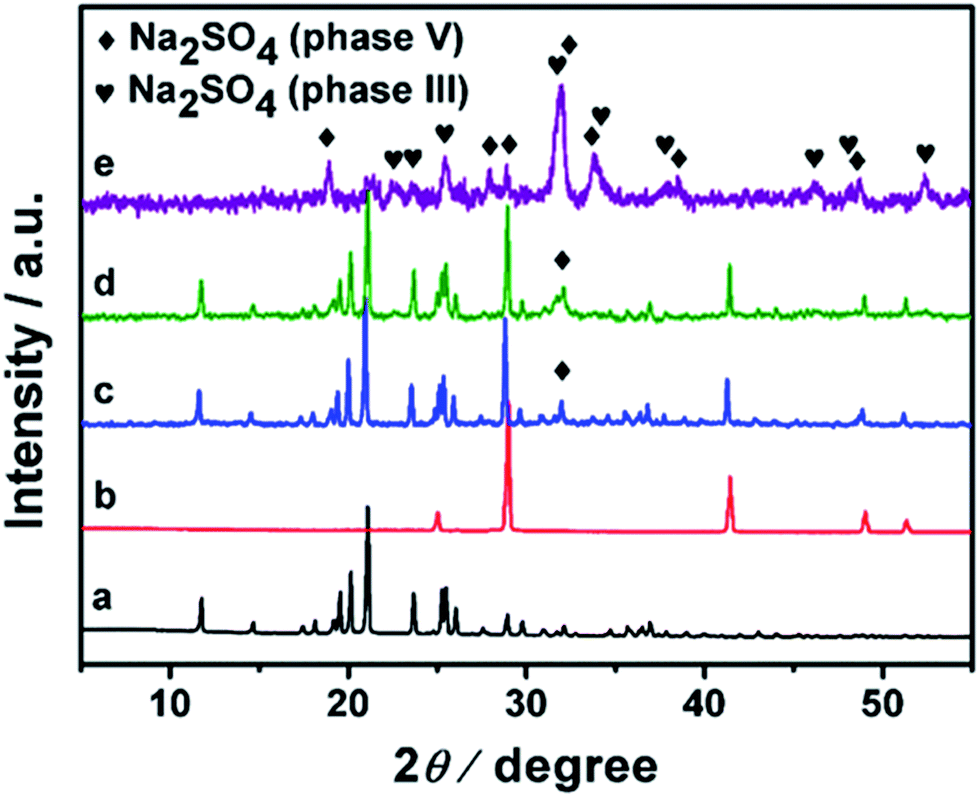 | ||
| Fig. 2 XRD patterns of (a) (Im-NH2)2SO4, (b) NaBH4 and (c–e) the samples after ball milling (Im-NH2)2SO4 and NaBH4 with different rotating speeds ((c) 100 rpm; (d) 150 rpm; (e) 200 rpm). | ||
Im-NH2-RT is further characterized by 11B and 13C SSNMR spectra (Fig. 3). The main peak of 11B SSNMR spectrum at −40.6 ppm and the main peaks of 13C SSNMR spectrum at 149.8 ppm, 146.2 ppm, 118.6 ppm and 109.6 ppm are assigned to BH4− and Im-NH2+ respectively, which are in accordance with previous studies of NaBH4 (ref. 31 and 45) and (Im-NH2)2SO4.46,47 The existence of the BH4− group is further evidenced by the FT-IR spectrum, in which characteristic B–H stretching bands at 2392 cm−1, 2295 cm−1 and 2229 cm−1 are clearly observed (Fig. S2†).
 | ||
| Fig. 3 11B SSNMR spectrum (a) and 13C SSNMR spectrum (b) of Im-NH2-RT. | ||
All of these results suggest that Im-NH2-RT contains BH4− and Im-NH2+. Considering the fact that Na2SO4 appeared and most NaBH4 and (Im-NH2)2SO4 are consumed as suggested by XRD, it is quite reasonable to conclude that Im-NH2BH4 are formed from the metathesis of NaBH4 and (Im-NH2)2SO4 by ball milling:
| 2NaBH4 + (Im-NH2)2SO4 → 2Im-NH2BH4 + Na2SO4. |
We also note some impurities in Im-NH2-RT, as indicated by the peaks in the 11B SSNMR spectrum at −23.1 ppm and the peaks of 13C SSNMR spectrum at 70.4 ppm, 39.2 ppm, 30.3 ppm. The peak of 11B SSNMR spectrum at −23.1 ppm is especially strong, which may be ascribed to a N–BH3 species according to the previous studies.37,40 In the case of GuaBH4, signals between −16 ppm and −24 ppm were assigned to the species with several guanidinium units containing terminal BH3 groups with different chemical environments.37 Hence, this N–BH3 species is very likely to form from the partial dehydrogenation of Im-NH2BH4 to C3N3H5BH3 during the ball milling process. The broad shoulder peak in the left of the peak at −23.1 ppm may be assigned to some more complex species such as (C3N3H5)2BH2 and (C3N3H5)3BH. The weak peaks of 13C SSNMR spectrum at 70.4 ppm, 39.2 ppm, 30.3 ppm imply that there may be sp3-C species in the Im-NH2-RT. It is possible because of the strong reducibility of BH4−. C–H bonds was also formed in the dehydrogenated products of GuaB3H8.40 Furthermore, the shoulder peak from 3000 cm−1 to 2890 cm−1 in the FT-IR spectrum also indicates the existence of sp3 C–H bonds (Fig. S2†).
We spent considerable efforts to avoid the by-products by optimizing the conditions of ball milling. Unfortunately, no significant improvement was obtained. Separation Im-NH2BH4 from the Im-NH2-RT was also attempted using anhydrous tetrahydrofuran (THF), acetonitrile and isopropanol. However, the extraction failed to yield sufficient product. Additionally, in the 1H NMR spectra of the liquid extract, no BH4− signals were observed (Fig. S3†). Using liquid ammonia as solvent to synthesize Im-NH2BH4 also failed (more details are available in the ESI†). For now, we will proceed to study the dehydrogenation properties of Im-NH2BH4 using Im-NH2-RT. Considering the high thermal stability of Na2SO4,43,44 it is reasonable to consider that its effect on the dehydrogenation of Im-NH2BH4 may be little.
Dehydrogenation properties of Im-NH2BH4
Fig. 4 shows the dehydrogenation profile of Im-NH2-RT. The TPD/MS curves (Fig. 4a) show that the onset dehydrogenation temperature is about 50.5 °C and there are three main hydrogenation peaks at 76.7 °C, 169.4 °C and 231.6 °C, respectively. No gaseous impurities were detected below 320 °C, which suggests that the strategy of balancing the number of H+ and H− to suppress ammonia release might be effective. In contrast, ammonia is not negligible during the dehydrogenation of GuaBH4. In addition, the onset dehydrogenation temperature and peak temperature of GuaBH4 are 90 °C and 100 °C respectively, both of which are higher than the corresponding temperature of Im-NH2BH4.38,41,48 This result suggests that combination of the charge transfer effect between BH4− and a large conjugated cation (Im-NH2+) and the interaction between H+ and H− indeed results in lower dehydrogenation temperature and also improved hydrogen purity. | ||
| Fig. 4 (a) TPD/MS curves of Im-NH2-RT. The heating rate is 5 °C min−1. The blue dotted line indicates the quantities of hydrogen released based on the total mass of sample, which calculated from the TCD integral area. (b) The dehydrogenation kinetics curve of Im-NH2-RT below 320 °C. | ||
The dehydrogenation capacity of each stage is 0.8 wt%, 0.9 wt% and 1.4 wt% respectively. The total quantities of hydrogen released is 3.1 wt% below 320 °C. Above 320 °C, some gaseous impurities are emitted which are most likely to derive from the decomposition of Im-NH2+. Therefore, we only focused on the hydrogenation properties below 320 °C.
Study on the dehydrogenation kinetics up to 320 °C (Fig. 4b) manifests that Im-NH2-RT can rapidly release 1.7 wt% hydrogen in the first 10 s and 3.0 wt% hydrogen in 20 min. At last, it can release 3.2 wt% hydrogen, which is in very good agreement with the TCD results. If the weight contribution of Na2SO4 is excluded, the hydrogen released will be 5.5 wt%. In other words, 2.8 equiv. H2 was released from Im-NH2BH4 below 320 °C. Considering the partial decomposition during the ball milling process, it is suggested that Im-NH2BH4 can release 3 equiv. H2 below 320 °C and approximately 1 equiv. H2 in every stage.
The dehydrogenation products and process of Im-NH2BH4
After dehydrogenation at 320 °C, the sample turned black, which is denoted as Im-NH2-320. Im-NH2-320 was also characterized by XRD (Fig. 5), SSNMR (Fig. 6) and FT-IR (Fig. S2†). The XRD patterns shows that the peaks are mainly ascribed to phase III and phase II of Na2SO4.44 The peak at 28.9° is assigned to NaBH4, which also exists in Im-NH2-RT. However, the assignment of the peak at 47.1° (marked by star) remains unsolved, which may be from the decomposition products of Im-NH2BH4. | ||
| Fig. 5 XRD pattern of Im-NH2-RT. | ||
 | ||
| Fig. 6 11B SSNMR spectrum (a) and 13C SSNMR spectrum (b) of Im-NH2-320. | ||
The main peak in 11B SSNMR spectrum of Im-NH2-320 at −40.6 ppm is indicative of BH4−, which is originated from the residual NaBH4 in Im-NH2-RT. This is in agreement with the high thermal stability of NaBH4, which remains stable up to 320 °C. The B–H stretching bands of BH4− are also observed in FT-IR spectrum (Fig. S2†). The peaks at 13.5 ppm and 0.6 ppm may correspond to 3 fold coordinated B species such as –BN3 and/or –BN2H.37,40 The peaks in 13C SSNMR spectrum at 161.9 ppm, 149.9 ppm and 15.6 ppm can be ascribed to the carbon atoms in the imidazole rings with different chemical environments compared to that of Im-NH2BH4. The peak at 42.7 ppm suggests formation of sp3 type carbon species. However, the characteristic signals of sp3-C–H disappear in the FT-IR spectrum of Im-NH2-320 (Fig. S2†), indicating the formation of C–B or/and C–N bonds. The change of the imidazole ring after dehydrogenation is expected, as release 3 equiv. H2 requires removing at least one H on the aromatic N.
Fig. 7 shows the DSC curve of Im-NH2-320 below 320 °C, in which there are three exothermic peaks at 96 °C, 186 °C and 261 °C. This matches well with the three-stage hydrogenation of Im-NH2BH4 measured by TPD/MS. The enthalpies of the three stages calculated from the DSC curve are −30.2 kJ mol−1, −28.3 kJ mol−1 and −19.8 kJ mol−1, respectively. Because of the endothermic effect of phase transformations of Na2SO4 from 200 °C to 280 °C,44 the virtual enthalpies of the latter two stages should be more negative.
 | ||
| Fig. 7 DSC curve of Im-NH2-RT. The heating rate is 5 °C min−1. | ||
There are four protonic H on N and four hydridic H on B atoms in Im-NH2BH4. The strong interaction between H+ and H− results in the exothermic hydrogenation process at low temperature, which has been observed in other B–N–H hydrogen storage systems, such as GuaBH4 and type compounds.38,49 Compared to GuaBH4, Im-NH2BH4 does not release ammonia during decomposition process as there are no excessive H+ in Im-NH2BH4. Furthermore, destabilization of BH4− by the large conjugated cation, Im-NH2+ effectively reduces the dehydrogenation temperature of Im-NH2BH4.
Based on the measured hydrogen capacity and the structural characterization of the dehydrogenated products, the dehydrogenation process of Im-NH2BH4 could be expressed as:
| Im-NH2BH4 → C3N3H3BH + 3H2. |
Although the above conclusion is obtained using a mixture of Im-NH2BH4 and Na2SO4, we believe it is indicative of most of the dehydrogenation properties of Im-NH2BH4, as Na2SO4 should be inert during the dehydrogenation process. We will continue to purify Im-NH2BH4 to test this hypothesis. With an attainable hydrogen capacity of 5.5 wt% and high hydrogen purity, Im-NH2BH4 will be a highly attractive hydrogen carrier.
Conclusions
In summary, Im-NH2BH4 can be synthesized by the metathesis of NaBH4 and (Im-NH2)2SO4 using ball milling. The formation of Im-NH2BH4 is identified by XRD, SSNMR and FT-IT spectra, albeit with a low purity. Im-NH2BH4 starts to release hydrogen at 50 °C, and can release 3.2 wt% pure hydrogen (Na2SO4 included) by three exothermic steps below 320 °C. Im-NH2BH4 has a theoretical hydrogen capacity of 8.1 wt%, therefore it releases 68% of the stored hydrogen below 320 °C. We believe the interaction between BH4− and Im-NH2+ and the interaction between H+ and H− accounts for the low dehydrogenation temperature of Im-NH2BH4, which corresponds with the initial expectation.Acknowledgements
The authors acknowledge the financial support from National Science Foundation of China (NSFC, No. U1201241, 11375020, 51431001 and 21321001).Notes and references
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- S. I. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS PubMed.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- Q. Lai, M. Paskevicius, D. A. Sheppard, C. E. Buckley, A. W. Thornton, M. R. Hill, Q. Gu, J. Mao, Z. Huang, H. K. Liu, Z. Guo, A. Banerjee, S. Chakraborty, R. Ahuja and K.-F. Aguey-Zinsou, ChemSusChem, 2015, 8, 2789–2825 CrossRef CAS PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- C. J. Webb, J. Phys. Chem. Solids, 2015, 84, 96–106 CrossRef CAS.
- K. F. Aguey-Zinsou and J. R. Ares-Fernandez, Energy Environ. Sci., 2010, 3, 526–543 CAS.
- R. Y. Yu, Q. A. Li and K. J. Li, Rare Met. Mater. Eng., 2007, 36, 1124–1128 CAS.
- D. L. Zhao and Y. H. Zhang, Rare Met., 2014, 33, 499–510 CrossRef CAS.
- W. Liu, C. J. Webb and E. M. Gray, Int. J. Hydrogen Energy, 2016, 41, 3485–3507 CrossRef CAS.
- F. A. H. Yap, N. S. Mustafa and M. Ismail, RSC Adv., 2015, 5, 9255–9260 RSC.
- J. F. Mao and D. H. Gregory, Energies, 2015, 8, 430–453 CrossRef CAS.
- E. Ronnebro, Curr. Opin. Solid State Mater. Sci., 2011, 15, 44–51 CrossRef.
- X. Liu, C. Wu, F. Wu and Y. Bai, Progr. Chem., 2015, 27, 1167–1181 Search PubMed.
- W. Cai, H. Wang, D. Sun, Q. Zhang, X. Yao and M. Zhu, RSC Adv., 2014, 4, 3082–3089 RSC.
- R. Moury and U. B. Demirci, Energies, 2015, 8, 3118–3141 CrossRef CAS.
- Z. H. Lu, Q. Yao, Z. J. Zhang, Y. W. Yang and X. S. Chen, J. Nanomater., 2014, 1, 11 Search PubMed.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 CAS.
- Y. H. Lu and Y. P. Feng, Nanoscale, 2011, 3, 2444–2453 RSC.
- L. F. Wang and R. T. Yang, Catal. Rev.: Sci. Eng., 2010, 52, 411–461 CAS.
- J. W. Ren, H. W. Langmi, B. C. North and M. Mathe, Int. J. Energy Res., 2015, 39, 607–620 CrossRef CAS.
- S. Proch, J. Herrmannsdorfer, R. Kempe, C. Kern, A. Jess, L. Seyfarth and J. Senker, Chem.–Eur. J., 2008, 14, 8204–8212 CrossRef CAS PubMed.
- Q. L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 CAS.
- T. He, Q. J. Pei and P. Chen, J. Energy Chem., 2015, 24, 587–594 CrossRef.
- C. Chung, J. Ihm and H. Lee, J. Korean Phys. Soc., 2015, 66, 1649–1655 CrossRef CAS.
- D. Bao, P. Gao, C. Li, G. Wu, Y. Wang, Y. Chen, H. Zhou and P. Yang, Eur. J. Inorg. Chem., 2016, 2016, 3371–3375 CrossRef CAS.
- H. W. Li, Y. G. Yan, S. Orimo, A. Zuttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- H. Kou, G. Sang, Y. Zhou, X. Wang, Z. Huang, W. Luo, L. Chen, X. Xiao, G. Yang and C. Hu, Int. J. Hydrogen Energy, 2014, 39, 11675–11682 CrossRef CAS.
- H.-W. Li, D. Matsumura, Y. Nishihata, E. Akiba and S.-I. Orimo, J. Jpn. Inst. Met., 2013, 77, 627–630 CrossRef CAS.
- G. Tu, X. Xiao, T. Qin, Y. Jiang, S. Li, H. Ge and L. Chen, RSC Adv., 2015, 5, 51110–51115 RSC.
- D. Ravnsbaek, Y. Filinchuk, Y. Cerenius, H. J. Jakobsen, F. Besenbacher, J. Skibsted and T. R. Jensen, Angew. Chem., 2009, 48, 6659–6663 CrossRef CAS PubMed.
- T. Jaron, P. A. Orlowski, W. Wegner, K. J. Fijalkowski, P. J. Leszczynski and W. Grochala, Angew. Chem., Int. Ed., 2015, 54, 1236–1239 CrossRef CAS PubMed.
- Q. Gu, Z. Wang, Y. Filinchuk, J. A. Kimpton, H. E. A. Brand, Q. Li and X. Yu, J. Phys. Chem. C, 2016, 120, 10192–10198 CAS.
- L. Liu, G. Wu, W. Chen, Z. Xiong, T. He and P. Chen, Int. J. Hydrogen Energy, 2015, 40, 429–434 CrossRef CAS.
- X. Zheng, X. Huang, Y. Song, X. Ma and Y. Guo, RSC Adv., 2015, 5, 105618–105621 RSC.
- H. Fu, Y. Wu, J. Chen, X. Wang, J. Zheng and X. Li, Inorg. Chem. Front., 2016, 3, 1137–1145 RSC.
- A. Doroodian, J. E. Dengler, A. Genest, N. Roesch and B. Rieger, Angew. Chem., Int. Ed., 2010, 49, 1871–1873 CrossRef CAS PubMed.
- T. J. Groshens and R. A. Hollins, Chem. Commun., 2009, 3089–3091, 10.1039/b900376b.
- G. Qi, K. Wang, X. Li and B. Zou, J. Phys. Chem. C, 2016, 120, 13414–13420 CAS.
- W. Chen, Z. Huang, G. Wu, T. He, Z. Li, J. Chen, Z. Guo, H. Liu and P. Chen, J. Mater. Chem. A, 2015, 3, 11411–11416 CAS.
- Z. Tang, Y. Guo, S. Li and X. Yu, J. Phys. Chem. C, 2011, 115, 3188–3193 CAS.
- G. Yanhui, G. Qinfen, G. Zaiping, M. Jianfeng, L. Huakun, D. Shixue and Y. Xuebin, J. Mater. Chem., 2011, 21, 7138–7144 RSC.
- B.-K. Choi and D. J. Lockwood, J. Phys.: Condens. Matter, 2005, 17, 6095–6108 CrossRef CAS.
- S. E. Rasmussen, J. E. Jorgensen and B. Lundtoft, J. Appl. Crystallogr., 1996, 29, 42–47 CrossRef CAS.
- R. W. Schurko, I. Hung, S. Schauff, C. L. B. Macdonald and A. H. Cowley, J. Phys. Chem. A, 2002, 106, 10096–10107 CrossRef CAS.
- Y. Zhang, J. Labelled Compd. Radiopharm., 2002, 45, 1055–1064 CrossRef CAS.
- F. Wilde, C. Chamseddin, H. Lemmerhirt, P. J. Bednarski, T. Jira and A. Link, Arch. Pharm., 2014, 347, 153–160 CrossRef CAS PubMed.
- Y. Guo, Q. Gu, Z. Guo, J. Mao, H. Liu, S. Dou and X. Yu, J. Mater. Chem., 2011, 21, 7138–7144 RSC.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and some experimental details. See DOI: 10.1039/c6ra21335a |
| This journal is © The Royal Society of Chemistry 2016 |