DOI:
10.1039/C6RA21284K
(Paper)
RSC Adv., 2016,
6, 97352-97362
Controllable synthesis of hierarchical NiCo2S4@Ni3S2 core–shell nanotube arrays with excellent electrochemical performance for aqueous asymmetric supercapacitors†
Received
24th August 2016
, Accepted 29th September 2016
First published on 29th September 2016
Abstract
In this study, unique NiCo2S4@Ni3S2 core–shell nanotube arrays (NTAs), a promising positive electrodes for supercapacitors, have been successfully synthesized on Ni foam via a novel method. Electrochemical tests show the highest area specific capacity of 4.25 C cm−2 at 4 mA cm−2, maintained at 3.12 C cm−2 at 40 mA cm−2. In addition, a 3D reduced graphene oxide (rGO) aerogel has been fabricated as a negative electrode for supercapacitors, and this displays an excellent capacitance performance of 286.9 C g−1 at 1 A g−1. An asymmetric supercapacitor denoted as NiCo2S4@Ni3S2//rGO has been assembled based on NiCo2S4@Ni3S2 core–shell NTAs and rGO aerogel. The NiCo2S4@Ni3S2//rGO device achieves an outstanding performance with a specific capacity of 163.15 C g−1, an energy density of 32.75 W h kg−1 at a power density of 0.36 kW kg−1. Moreover, it displays a remarkable cycling performance (77.5% capacity retention after 5000 cycles). These results indicate potential applications of NiCo2S4@Ni3S2//rGO in asymmetric supercapacitors (ASCs).
1. Introduction
In response to environmental problems and the energy crisis, the development of environmentally-friendly and renewable sources of energy has become an urgent worldwide necessity. Supercapacitors, also called electrochemical capacitors, have attracted intense interest due to their high power density, long cycle life, fast charge–discharge capability, and excellent reversibility.1–3 Early research mainly focused on electrical double-layer capacitors (EDLCs), such as porous carbons and graphenes, whose capacity stems from the interface between electrolyte and electrode.4,5 Nevertheless, their low energy density and low specific capacity greatly limit their application in energy storage devices.6,7 Currently, redox-based supercapacitors have shown potential applications for energy storage devices owing to their high specific capacity and reversible redox reaction.8,9 Universally, faradaic materials are mainly transition metal oxides,10–12 transition metal sulfides,13,14 transition metal nitrides,15,16 transition metal phosphides,17,18 hydroxides,19,20 and conductive polymers.21,22 Although these supercapacitive materials can display a remarkably high faradaic capacity compared to carbon materials resulting from a reversible redox reaction, there is an urgent problem to be addressed by researchers: relatively low energy density hinders the promotion and application of supercapacitors. Lately, numerous researchers have begun to develop ASCs due to their high energy density and high power density on account of their high operating voltage and high specific capacity.23,24 Hence, we usually fabricate ASCs by the combination of faradaic and EDLC materials to accomplish high power density, high energy density and a stable cycling life.25 Considering the above, it is critical to seek a prominent faradaic material and an outstanding EDLC material in order to boost the performance of ASCs.
Recently, metal sulfides have emerged as one of the most outstanding candidates for supercapacitors, such as binary nickel sulfides,26,27 cobalt sulfides28 and manganese sulfides.29 Compared to the low capacity of carbon materials, the poor electrical conductivity of transition metal oxides, and the poor cycle performance of conducting polymers, transition metal sulfides have displayed numerous superior electrochemical properties.30 Among metal sulfides, NiCo2S4 possesses richer redox reactions than the corresponding binary nickel sulphide (NiS) and cobalt sulphide (Co9S8), and exhibits a major superiority over NiCo2O4 in terms of higher conductivity and higher specific capacity.31,32 In fact, there have been some significant achievements in the application to supercapacitors of NiCo2S4 electrode materials.33,34 For instance, Wu et al. assembled an ASC using NiCo2S4 mesoporous nanosheets as the positive electrode and active carbon (AC) as the negative electrode. This ASC exhibited an energy density of 25.5 W h kg−1 at a power density of 334 W kg−1.35 However, single NiCo2S4 electrode materials cannot meet the requirements of supercapacitors for high power density and high energy density. The facile and recommended strategy to conquer this defect is to construct a core–shell structure with different supercapacitive materials, where the differences are due to their rich accessible redox reaction sites, easy diffusion of electrolyte, enlarged contact surface area between electrode and electrolyte, as well as the fantastic synergy effect between core and shell materials. There have been some reports on constructing core–shell structures that have achieved supercapacitors with superior electrochemical performances. Fu et al. successfully fabricated hierarchical NiCo2S4@CoSx hybrid electrodes by a two-step method, which show an excellent specific capacity of 2.37 C cm−2 at 5 mA cm−2.36 Wan et al. constructed NiCo2S4 nanotube@NiMn-LDH arrays, which show a high specific capacity of 0.87 C cm−2 at 1 mA cm−2.37 Evidently, NiCo2S4-based hybrid core–shell structures have proved to be promising positive electrodes in applications to make high performance ASCs.
As for the negative electrode materials in ASCs, 3D porous rGO aerogels have lately been considered as ideal EDLC materials for high-performance supercapacitors, because of their superior properties like large surface areas, excellent cycle stability, high electrical conductivity and well-controlled structures.38–40 Moreover, 3D rGO aerogels can guarantee multi-dimensional electron transport pathways and ease of access to the electrolyte, which are of great importance for achieving high-rate energy storage.41 Therefore, 3D porous rGO aerogels can be considered as candidate negative electrodes for ASCs.
In this paper, we have controlled the synthesis of unique hierarchical NiCo2S4@Ni3S2 core–shell NTAs in situ grown on Ni foam and used as positive electrodes. Moreover, a 3D rGO aerogel with an excellent capacity performance was fabricated as a negative electrode through a facile hydrothermal reaction. First, a core material of NiCo2S4 NTAs was constructed via a facile and effective anion-exchange reaction. This offered a stable scaffold for the shell and a short diffusion path for electrolyte ions and abundant active sites for the faradaic process. Then ultrathin Ni3S2 nanosheets are coated on the surface of NiCo2S4 NTAs by a simple electrodeposition process. To the best of our knowledge, the enlarged surface of the Ni3S2 nanosheets is favorable for the rapid diffusion of electrolyte ions and electrolyte efficient infiltration.42,43 Remarkably, the NiCo2S4@Ni3S2 core–shell NTAs as-fabricated through the combination of NiCo2S4 nanotubes and Ni3S2 nanosheets exhibited a superior electrochemical performance as a supercapacitor, which could be ascribed to the synergistic effect of its unique core–shell nanostructures. Furthermore, the as-assembled NiCo2S4@Ni3S2//rGO device exhibits relatively high energy density, high power density and excellent cycling stability, indicating its great potential in ASCs.
2. Experimental sections
2.1. Synthesis of NiCo2S4 NTAs on Ni foam
Unique NiCo2S4 NTAs were synthesized according to previously published work.44 Prior to the synthesis, the Ni foam (1 cm × 1 cm × 1.6 mm) was pretreated with 5% HCl, ethanol and deionized (DI) water for 15 min each to remove impurities and oxides from the surface and dried in an oven. NiCo2S4 NTAs were obtained through a two-step hydrothermal reaction. Firstly, 2 mmol (0.4754 g) NiCl2·6H2O, 4 mmol (0.9517 g) CoCl2·6H2O and 12 mmol (0.7207 g) urea were dissolved in 35 mL DI water with continuous stirring. Then the solution was transferred into a 50 mL Teflon-lined stainless steel autoclave. Subsequently, we strung six nickel foams by using a bent iron wire and put them in the center of the Teflon. The autoclave was kept at 120 °C for 6 h in the oven and cooled down to room temperature naturally. The samples were collected by washing with DI water and ethanol, then dried in the oven at 60 °C for 12 h to make precursors. Secondly, the precursors were placed in a 50 mL Teflon-lined stainless steel autoclave with 35 mL sodium sulfide solution (0.2 M) and kept at 120 °C for 14 h. Finally, the products were collected by washing with DI water and ethanol, and then dried at 60 °C for 12 h to obtain NiCo2S4 NTAs. The mass loading of NiCo2S4 on Ni foam was approximately 8.0 mg.
2.2. Synthesis of NiCo2S4@Ni3S2 core–shell NTAs
Ni3S2 nanosheets were synthesized via an electrodeposition process carried out on a CHI630D electrochemical analyzer.45 Including a Ni foam sheet supported NiCo2S4 NTAs as the working electrode, a saturated Ag/AgCl as the reference electrode and a Pt sheet (1 cm × 1 cm) as the counter electrode. The typical deposition was formed in a mixed solution of 50 mM NiCl2·6H2O and 1 M thiourea by cyclic voltammetry (CV) with the potential ranging from −1.2 V to 0.2 V (vs. Ag/AgCl) at 5 mV s−1 for 6 cycles. The NiCo2S4@Ni3S2 core–shell NTAs were collected by washing with DI water and ethanol, then dried at 60 °C for 12 h. As a comparison, bare Ni3S2 was obtained through the same synthesis method, except that the working electrode was the clean Ni foam. The mass loading of Ni3S2 was about 2.0 mg and the whole weight of NiCo2S4@Ni3S2 core–shell NTAs on Ni foam was 10.0 mg.
2.3. Preparation of the 3D rGO aerogel
A graphene oxide (GO) solution was initially prepared from natural graphite by a modified Hummers' method.46 The obtained GO solution was diluted to 2 mg mL−1. Then 70 mL of the diluted GO solution was added to a 100 mL enclosed Teflon-lined autoclave and maintained at 180 °C for 12 h. The autoclave was cooled to room temperature naturally. A self-assembled columnar graphene was formed and vacuum-freeze-dried for 24 h to obtain the 3D rGO aerogel.
2.4. Characterization
Crystallite structures of the as-prepared samples were characterized by X-ray diffraction (XRD, Rigaku Smart Lab) using Cu Kα (λ = 1.5418 Å) radiation. The morphology and microstructure of the synthesized products were examined by field emission scanning electron microscopy (FESEM, SU-8000, Japan) and high resolution transmission electron microscopy (HRTEM, JEM-2100). Raman spectra were obtained using a micro-Raman system (LabRAM HR800) with an excitation energy of 2.41 eV (514 nm).
2.5. Electrochemical measurements
The electrochemical performances of the obtained samples were investigated in a three-electrode cell in a 2 M KOH aqueous solution. The obtained samples were used as the working electrode, a platinum plate (3 cm × 3 cm) was used as the counter electrode and Hg/HgO was used as the reference electrode. Cyclic voltammograms (CV), galvanostatic charge–discharge (GCD) measurements were carried out on an electrochemical workstation (CHI660E) to evaluate the electrochemical behaviors. And the long cycling life of the ASC devices was inspected through a LAND battery test system (CT2001A). The average specific capacity determined from galvanostatic charge–discharge (GCD) tests can be calculated from eqn (1) as follows:| |
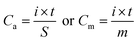 | (1) |
here Ca is the area specific capacity (C cm−2), Cm is the mass specific capacity (C g−1), i is the discharge current (A), t is the discharge time (s), S is the surface area (cm−2) and m is the mass (g) of a single electrode.
2.6. Fabrication of ASC devices
The ASC devices were fabricated in a two-electrode configuration by taking NiCo2S4@Ni3S2 NTAs as the positive electrode and rGO aerogel as the negative electrode with a separator made by polyvinylidene fluoride (PVDF) sandwiched between the two electrodes. Prior to the assembly of the ASC devices, the rGO electrode was obtained by mixing rGO, polytetrafluorene-ethylene (PTFE) and acetylene black in ethanol with a mass ratio (%) of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. The prepared mixture was ultrasonicated for 1 h and coated on Ni foam according to the matched weight. Afterwards, the Ni foam with the rGO coating was pressed and dried at 60 °C for at least 12 h.
:10:10. The prepared mixture was ultrasonicated for 1 h and coated on Ni foam according to the matched weight. Afterwards, the Ni foam with the rGO coating was pressed and dried at 60 °C for at least 12 h.
3. Results and discussion
3.1. Structural characterization of NiCo2S4@Ni3S2 core–shell NTAs
Hydrothermal synthesis combined with an electrodeposition method were used for the hierarchical NiCo2S4@Ni3S2 core–shell NTAs on Ni foam, which is schematically illustrated in Fig. 1. Initially, aligned NiCo2S4 NTAs were grown on Ni foam by a two-step hydrothermal method. Subsequently, the NiCo2S4 NTAs served as the scaffold for the growth of interconnected Ni3S2 ultrathin nanosheets via an electrodeposition process and formed unique hierarchical core–shell hybrid structures. The hierarchical NiCo2S4 NTAs on Ni foam are shown in Fig. 2 at different magnifications. In Fig. 2(a), the Ni foam is covered with a large number of NiCo2S4 NTAs. Obviously, the hierarchical structures adhere to the Ni foam substrate tightly and uniformly. The magnified image (Fig. 2(b)) reveals that the NiCo2S4 nanotubes have diameters ranging from 50 to 150 nm and lengths of 2–4 μm from their tips to their roots in contact with the Ni foam. The unique morphology and structure of NiCo2S4 nanotubes can be attributed to the process of the anion-exchange reaction during the conversion from precursors to NiCo2S4.47,48
 |
| | Fig. 1 Schematic illustration of a facile method of NiCo2S4@Ni3S2 core–shell NTAs on Ni foam. | |
 |
| | Fig. 2 FESEM images of (a) and (b) NiCo2S4 NTAs on Ni foam. | |
For the sake of a closer exploration of the morphologies of the NiCo2S4@Ni3S2 core–shell NTAs, a series of parallel experiments were carried out. The NiCo2S4 nanotubes covered with slim Ni3S2 nanosheets with different CV cycles of electrodeposition are displayed in Fig. 3. As shown in Fig. 3(a), Ni3S2 nanosheets can be observed; the core–shell structures are not very evident. This results from the small amount of Ni3S2 with just three deposition cycles. In Fig. 3(b), after nine cycles, the NiCo2S4 nanotubes are not only covered by a large number of Ni3S2 nanosheets, but there also appear to be vast agglomerations of particles, leading to less space between the core–shell nanotubes. In terms of experimental parameters, the optimal morphologies are obtained after six cycles, as shown in Fig. 3(c) and (d). The Ni3S2 nanosheets are interconnected with each other, thus forming a highly porous surface morphology. The morphology of Ni3S2 was also characterized by FESEM (Fig. S1†). In addition, the spaces between each core–shell nanotube constitute ordered channels on a large scale. In such a unique core–shell structure, the opened channels served as diffusion pathways, making the active materials more accessible to electrolyte ions.41 Moreover, those ultrathin and curled Ni3S2 nanosheets greatly increase the redox sites during electrochemical behaviours.49
 |
| | Fig. 3 FESEM images of (a) NiCo2S4 NTAs covered by three cycles Ni3S2 nanosheets; (b) NiCo2S4 NTAs covered by nine cycles Ni3S2 nanosheets; (c) and (d) NiCo2S4 NTAs covered by six cycles Ni3S2 nanosheets. | |
The sample structures were further confirmed by XRD analysis. Fig. 4 shows the wide-angle XRD patterns of the NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs supported on the Ni foam. The three typical peaks at 44.7°, 52.1° and 76.5° originate from the Ni substrate concerned with the (111), (200) and (220) planes (JCPDS no. 87-0712).50 The five major peaks at 31.6°, 38.3°, 47.4°, 50.5°, and 55.3° were indexed to the (311), (400), (422), (511) and (440) planes of the NiCo2S4 phase (JCPDS card no. 20-0782). The EDX spectrum (Fig. S2(a)†) indicates that the NiCo2S4 nanotubes consist mainly of Ni, Co and S elements. Moreover, the weak peak at 21.8° coincides with the (101) plane in the standard Ni3S2 spectrum (JCPDS card no. 44-1418), indicating poor crystallinity or the low mass loading of Ni3S2 nanosheets obtained by electrodeposition. The chemical compositions of the Ni3S2 nanosheets were confirmed by EDX (Fig. S2(b)†). The results clearly reveal that the Ni3S2 nanosheets are composed mainly of Ni and S. The Ni/S atomic ratio is about 1.45, which indicates that the chemical composition of the Ni3S2 nanosheets is pure Ni3S2.
 |
| | Fig. 4 Powder XRD patterns of the NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs on Ni foam. | |
TEM analysis provides further insight into the morphology and detailed structure of the as-obtained NiCo2S4@Ni3S2 core–shell NTAs. The whole of the core–shell structures can be clearly observed, as shown in Fig. 5(a)–(c), while the core NiCo2S4 nanotubes are tightly wrapped by Ni3S2 nanosheets. Also the hollow part of the NiCo2S4 nanotube is filled up with the shell. As shown in Fig. 5(d) and (e), there are straight and orderly lattice fringes with interplanar gaps of 0.28 nm, corresponding well to the (311) plane of NiCo2S4. The other image with bending and relatively coarse lattice fringes represents an interplanar spacing of 0.41 nm which can be indexed to the (101) plane of Ni3S2, suggesting poor crystallinity compared with the NiCo2S4 nanotubes as well as in the XRD analysis.
 |
| | Fig. 5 (a–c) HRTEM images of NiCo2S4@Ni3S2 core–shell NTAs; (d) HRTEM images of Ni3S2 nanosheets; (e) HRTEM images of NiCo2S4 NTAs. | |
3.2. Electrochemical performance of the NiCo2S4@Ni3S2 core–shell NTAs
To explore the electrochemical performances of the samples, cyclic voltammograms (CV) measurements were performed on the three-electrode cell in 2 M KOH aqueous solution. Fig. 6(a) shows the CV curves of pure NiCo2S4 and NiCo2S4@Ni3S2 core–shell NTAs electrodes in the voltage range of 0–0.6 V (vs. Hg/HgO) at a scan rate of 5 mV s−1. The obvious redox peaks indicated the faradaic capacitive characteristics of the samples. For the NiCo2S4 NTAs electrode, a pair of redox peaks at 0.23 V and 0.48 V (Ni2+/Ni3+ and Co3+/Co4+) can be clearly observed. However, compared with the CV curve of bare NiCo2S4, the NiCo2S4@Ni3S2 core–shell NTAs electrode shows a pair of significantly enhanced redox peaks at voltages of 0.22 V and 0.51 V, respectively, which may be derived from the abundant faradaic redox reactions and the short ion diffusion path offered by Ni3S2, which can be attributed to the reversible reactions of Ni2+/Ni3+ caused by the addition of Ni3S2 nanosheets (the supercapacitor performance of Ni3S2 is supplied in Fig. S3†). Moreover, the area surrounded by the CV curve is dramatically enlarged by the introduction of Ni3S2 nanosheets, suggesting the larger capacity of NiCo2S4@Ni3S2 core–shell NTAs. Fig. 6(b) shows CV curves of NiCo2S4@Ni3S2 electrodes at 5–50 mV s−1. With an increase in the scan rate, the CV curves have a tendency to further augment the shape and shifting of the redox peaks, suggesting a diffusion-controlled electrochemical process. The charge storage mechanism of NiCo2S4 and NiCo2S4@Ni3S2 core–shell NTAs can be described by the following equations in alkaline electrolyte.40,51| | |
NiS + OH− ↔ NiSOH + e−
| (2) |
| | |
CoS + OH− ↔ CoSOH + e−
| (3) |
| | |
CoSOH + OH− ↔ CoSO + H2O + e−
| (4) |
| | |
Ni3S2 + 3OH− ↔ Ni3S2(OH)3 + 3e−
| (5) |
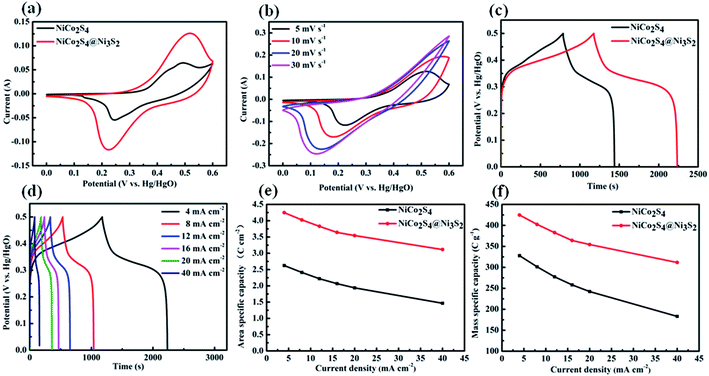 |
| | Fig. 6 (a) CV curves of the NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs at 5 mV s−1; (b) CV curves of NiCo2S4@Ni3S2 core–shell NTAs at various scan rates; (c) galvanostatic charge–discharge curves of NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs at 4 mA cm−2; (d) galvanostatic charge–discharge curves of NiCo2S4@Ni3S2 core–shell NTAs at different current densities; (e) and (f) Ca and Cm of NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs at different current densities. | |
Fig. 6(c) depicts a comparison of charge–discharge curves for the NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs between 0 and 0.5 V at a current density of 4 mA cm−2. As expected, the NiCo2S4@Ni3S2 core–shell NTAs electrode achieves a much longer charge–discharge time than the bare NiCo2S4, demonstrating its smaller polarization and higher electrochemical reactivity. According to eqn (1), the area specific capacity of the NiCo2S4@Ni3S2 electrode is calculated to be 4.25 C cm−2 (425 C g−1), which is evidently higher than the bare NiCo2S4 with 2.62 C cm−2 (328 C g−1) at the lowest current density of 4 mA cm−2. The charge–discharge curves of the NiCo2S4@Ni3S2 core–shell NTAs at various current densities are recorded in Fig. 6(d). Fig. 6(e) further illustrates the area specific capacity (Ca) of the NiCo2S4 NTAs and NiCo2S4@Ni3S2 core–shell NTAs at various current densities. Meanwhile, the mass specific capacity (Cm) based on the mass loading of all active materials versus current density plots are shown in Fig. 6(f). The specific capacities decrease gradually with increasing current density, because the ions don't have enough time to penetrate into the inner portion of the active materials at a high current density. Nevertheless, the areal specific capacity of NiCo2S4@Ni3S2 is still sustained at 3.12 C cm−2 at 40 mA cm−2 (capacity retention of 73.4% at 40 mA cm−2). Furthermore, the specific capacity and capacity retention of NiCo2S4@Ni3S2 electrode in this study are superior to numerous NiCo2S4-based nanomaterials reported before, such as NiCo2S4@CoSx (2.37 C cm−2 at 5 mA cm−2, capacity retention of 47.7% at 50 mA cm−2),36 NiCo2S4@Ni–Mn LDH arrays/GS (0.87 C cm−2 at 1 mA cm−2, capacity retention of 72.8% at 10 mA cm−2),41 NiCo2S4@MnO2 (1.30 C cm−2 at 3 mA cm−2, capacity retention of 46.2% at 20 mA cm−2).52
3.3. Structural characterization and electrochemical performance of 3D rGO aerogel
Obviously, the high-resolution SEM image in Fig. 7(a) indicates that rGO aerogel has an interconnected 3D porous network with pore sizes ranging from submicrometers to micrometers. This can be clearly observed in the TEM (Fig. 7(b)). The phase structures of rGO and GO were checked by XRD (Fig. S4(a)†). The curve of GO displays a peak at 2θ = 11.5°, which corresponds to an interlayer distance of 0.76 nm originating from some functional groups between the layers, such as carboxyl, hydroxyl, and epoxy. The curve of rGO shows a broad peak at 2θ = 24.4°, indicating its poor ordering characteristic. The changes in the structure were researched by Raman spectroscopy (Fig. S4(b)†). As shown in the figure, the peak for rGO (1589 cm−1) at the G band is down-shifted compared with that of GO (1593 cm−1), indicating that the hexagonal network of carbon atoms in rGO recovers with defects from the structure with rich isolated double bonds in GO.53,54 The ID/IG ratio of GO is 0.98 and increases to 1.10 in rGO, which demonstrates that some defects introduced in rGO result from hydrothermal reduction, which is related to the disordered carbon structure. Examples are the vacancies, boundaries, and amorphous structure, which can provide more active sites for charge storage and boost electrochemical performance. To discuss the pore structure of 3D rGO aerogel, a N2 adsorption–desorption measurement was carried out at 77 K. Fig. S5† gives the N2 adsorption–desorption isotherms of the 3D rGO aerogel, based on which the BET specific surface area is calculated to be 168.6 m2 g−1 (Table S1†). Pore size distribution analysis using the Barrett–Joyner–Halenda (BJH) method shows that the 3D rGO aerogel has a wide pore-size distribution, from micropores to macropores (inset of Fig. S5†), which is consistent with the FESEM observations.
 |
| | Fig. 7 (a) The high-resolution FESEM images of rGO aerogel; (b) the high-resolution TEM images of rGO aerogel; (c) CV curves of rGO aerogel at various scan rates; (d) galvanostatic charge–discharge curves of rGO aerogel at different current densities; (e) Cm of rGO aerogel and AC at different current densities; (f) long-term cycle life graph of rGO aerogel and AC. | |
The CV curves of the rGO aerogel show a similar rectangular shape at different scan rates, with a potential window of −1.0 to 0.0 V (vs. Hg/HgO) in a 2 M KOH electrolyte solution, as can be seen in Fig. 7(c), which reveals the typical double-layer capacity. The GCD curves of the rGO aerogel are relatively symmetrical and linear, which also indicates the double-layer capacitive behavior (Fig. 7(d)) of the rGO aerogel. Fig. 7(e) displays the calculations of the specific capacity for the rGO aerogel according to the galvanostatic discharge curves. The specific capacity for the rGO aerogel can achieve a value of 286.9 C g−1 at a current density of 1 A g−1, which is superior to AC (240.1 C g−1 at 1 A g−1). When the current density increases to 10 A g−1, the specific capacity of the rGO aerogel can also achieve 210.1 C g−1, manifesting an excellent property of rGO aerogel at a high discharge rate. The cycling performance of the rGO aerogel at a current density of 10 A g−1 is also shown in Fig. 7(f). The capacity retention can remain as high as 93.2% of the maximum value after 5000 cycles, showing the superior cycling stability of the 3D rGO aerogel. In contrast, the specific capacity retention of AC is only 83.6% under the same conditions. In sum, the rGO aerogels, which possess an interconnected 3D porous network exhibiting a symmetrical linear characteristic, demonstrate admirable EDLC properties, including outstanding cycle stability and excellent rate capability, due to their high specific surface areas, large pore volumes and superior electrical conductivity.
3.4. Electrochemical performances of the ASC device
To further investigate the electrochemical performances of the composite material, an ASC was fabricated by using NiCo2S4@Ni3S2 core–shell NTAs as the positive electrode and 3D rGO aerogel as the negative electrode, and NiCo2S4//rGO was assembled for comparison. For the two-electrode system, it is crucial to keep the charges balanced with the relationship q+ = q−.55 According to eqn (6) and (7):
According to the charge balance (q+ = q−), the mass of rGO was decided by following eqn (8):
| |
 | (8) |
here,
q+,
m+ and
Cm+ represent the charge (C), mass (g) and specific capacity (C g
−1) of the positive electrode;
q−,
m−, and
Csp− represent the charge (C), mass (g), and specific capacity (C g
−1) of the negative electrode in the three-electrode cell. After this calculation, the mass loadings of rGO in NiCo
2S
4//rGO and NiCo
2S
4@Ni
3S
2//rGO were 9.25 and 15.20 mg, respectively.
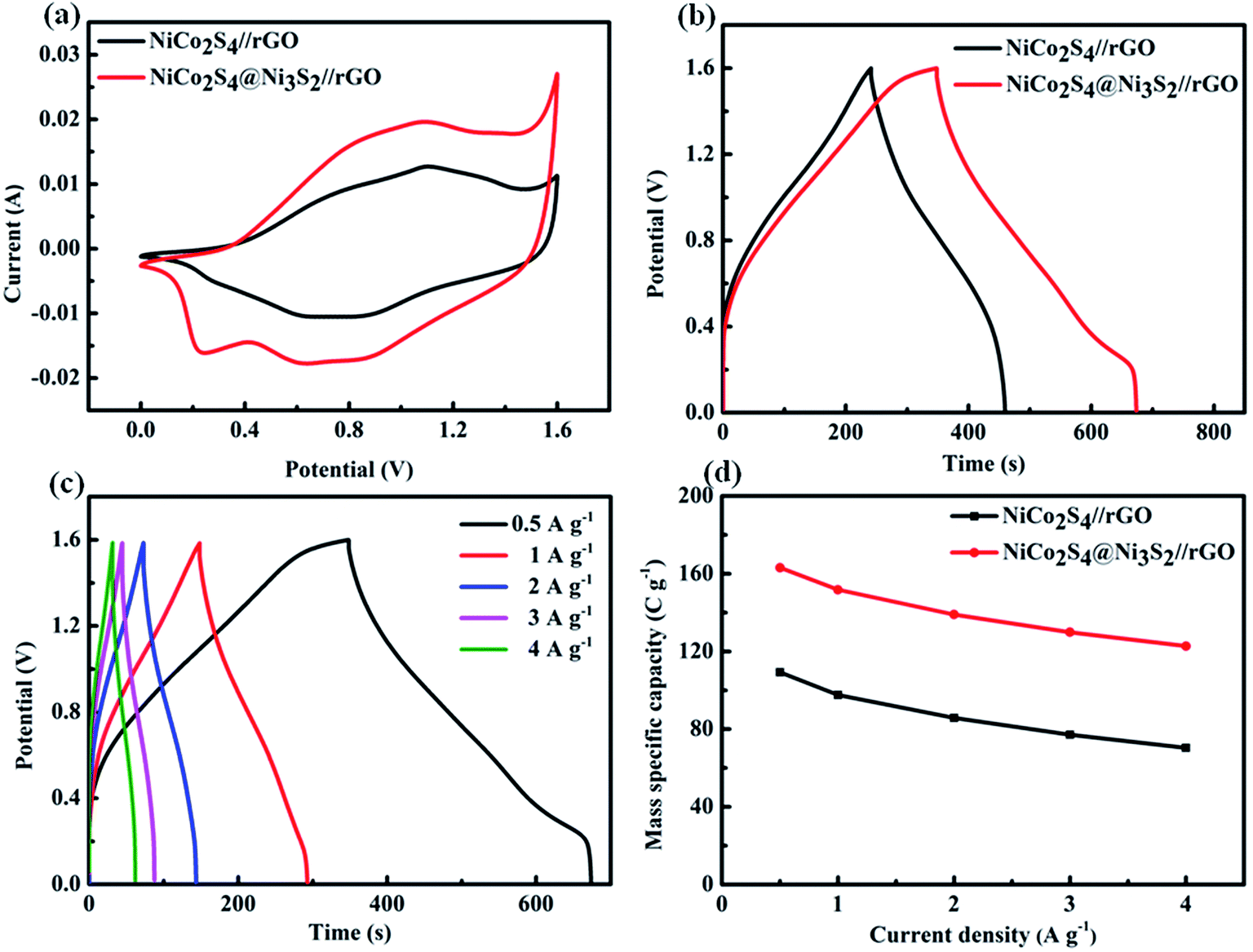
Fig. 8(a) exhibits the CV curves of NiCo
2S
4//rGO and NiCo
2S
4@Ni
3S
2//rGO at a scan rate of 5 mV s
−1 under 1.6 V in 2 KOH electrolyte. It is apparent that the area enclosed by the CV curves of the NiCo
2S
4@Ni
3S
2//rGO is considerably larger than that of the NiCo
2S
4//rGO device, which benefits from the construction of NiCo
2S
4@Ni
3S
2 core–shell structure. Galvanostatic charge–discharge tests were further conducted in the voltage range from 0 to 1.6 V to estimate the capacity of NiCo
2S
4@Ni
3S
2//rGO and NiCo
2S
4//rGO. As shown in
Fig. 8(b) and (c), the NiCo
2S
4@Ni
3S
2//rGO still shows a longer discharge time than bare NiCo
2S
4//rGO, indicating that the core–shell structure is endowed with stable and exceptional electrochemical performance both in an asymmetric supercapacitor and in the three-electrode system. Furthermore, the
Cm values of the NiCo
2S
4//rGO and NiCo
2S
4@Ni
3S
2//rGO devices depended on the discharging curves with different current densities, as plotted in
Fig. 8(d). Evidently, the mass specific capacity of NiCo
2S
4@Ni
3S
2//rGO achieves 163.15 C g
−1 at a current density of 0.5 A g
−1 and 122.80 C g
−1 at a high current density of 4 A g
−1.
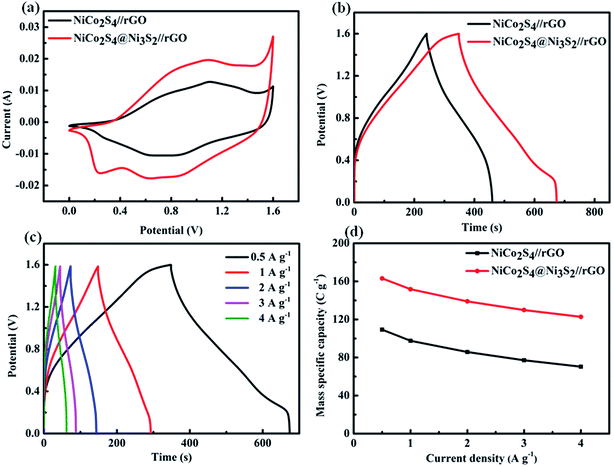 |
| | Fig. 8 (a) CV curves of the NiCo2S4//rGO and NiCo2S4@Ni3S2//rGO at 5 mV s−1; (b) galvanostatic charge–discharge curves of the NiCo2S4//rGO and NiCo2S4@Ni3S2//rGO at 0.5 A g−1; (c) galvanostatic charge–discharge curves of NiCo2S4@Ni3S2//rGO at different current densities; (d) Cm of NiCo2S4//rGO and NiCo2S4@Ni3S2//rGO at different current densities. | |
The cycling performances of NiCo2S4//rGO and NiCo2S4@Ni3S2//rGO were also investigated at a current density of 2 A g−1. As shown in Fig. 9(a), an obviously increasing capacity is captured owing to the activation process of the electrode materials for the first 500 cycles in the test.56,57 In particular, the mass loading of the ASC devices was so high that it took some time to activate the active materials. After reaching the maximum, both devices show a fluctuating decline, ending up with 77.6% and 69.4% of the initial capacity after 5000 cycles, respectively. The excellent cycling performance of NiCo2S4@Ni3S2//rGO with little deformation expresses a favourable stability due to the structure of NiCo2S4@Ni3S2 and the strong adherence to the substrate Ni foam. We have also provided the XRD pattern and FESEM image of NiCo2S4@Ni3S2 on the surface of the Ni foam after the 5000 cycling tests in Fig. S6 and S7.† The test results showed that the core–shell nanotube structure of NiCo2S4@Ni3S2 remains mainly stable, which also proved the long cycle life of the ACS device. Fig. S8† shows the impedance Nyquist plots of the NiCo2S4, Ni3S2 and NiCo2S4@Ni3S2 composite electrodes. Remarkably, the internal resistances (Re) at the high-frequency intercept of the real axis are measured to be 0.75, 0.46 and 0.32 Ω for NiCo2S4, NiCo2S4@Ni3S2 and Ni3S2, respectively. The NiCo2S4@Ni3S2 exhibits a slightly smaller semicircle than the Ni3S2 and NiCo2S4 electrode at the medium-frequency, which indicates the lower charge transfer resistance (Rct).58 NiCo2S4@Ni3S2 also reveals a higher slope at low frequency compared to the Ni3S2 and NiCo2S4 electrodes, demonstrating its lower diffusive resistance (W).59 The above are responsible for the excellent electrochemical performance of the NiCo2S4@Ni3S2 core–shell NTAs.
 |
| | Fig. 9 (a) long-term cycle life graph of NiCo2S4//rGO and NiCo2S4@Ni3S2//rGO; (b) Ragone plots of energy density and power density of NiCo2S4@Ni3S2//rGO. | |
To highlight the superior electrochemical performance of the ASC device, an advanced Ragone plot of NiCo2S4@Ni3S2//rGO (energy density vs. power density) is displayed in Fig. 9(b). The equations given below were used to calculate the energy density and power density of the ASC devices:
| |
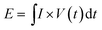 | (9) |
where
E is the energy density (W h kg
−1),
P is the power density (W kg
−1),
V is the cell voltage (V) and
t is the discharge time (s) of the ASC devices. It is worth noting that the NiCo
2S
4@Ni
3S
2//rGO device exhibits an outstanding energy density of 32.75 W h kg
−1 at a power density of 0.36 kW kg
−1 and still retains a considerable energy density of 25.50 W h kg
−1 at a large power density of 2.98 kW kg
−1, implying its feasible and promising applications for energy storage. Additionally, the obtained high energy and power densities of our supercapacitors are superior to the values reported so far among the ASC devices based on NiCo
2S
4, such as hollow NiCo
2S
4 (microspheres)//AC (24.7 W h kg
−1 at 0.428 kW kg
−1),
60 and mesoporous NiCo
2S
4 (nanoparticles)//AC (28.3 W h kg
−1 at 0.245 kW kg
−1).
61 Remarkably, a series of ASC devices based on core–shell structures are also displayed for comparison. The value of the NiCo
2S
4@Ni
3S
2//rGO device also possesses a competitive superiority compared with CNT@NiO//PCPs (25.4 W h kg
−1 at 0.4 kW kg
−1),
62 NiCo
2O
4@NiMoO
4//AC (21.7 W h kg
−1 at 0.157 kW kg
−1)
63 and so on. In order to investigate the practical application of NiCo
2S
4@Ni
3S
2//rGO, two ASC devices were assembled by series connection. After charging, the NiCo
2S
4@Ni
3S
2//rGO device is able to light up a red commercial light-emitting diode (LED) with a diameter of 5 mm (2.2 V, 20 mA) as shown in
Fig. 9(b).
Based on the above results, NiCo2S4@Ni3S2 core–shell NTAs electrodes possess an ultrahigh specific capacity, a superior rate capability and a fast electron transport and ion diffusion rate, because: (I) NiCo2S4 with intrinsic properties of good conductivity and high specific capacity is made into a hollow tubular structure, resulting in a sufficient connection between the electrolyte and the electrode which can significantly promote full utilization of the electrode;47,48,64 (II) the ultrathin interconnected Ni3S2 nanosheets are tightly wrapped on the surface of NiCo2S4 NTAs, contributing to enhanced mechanical stability and effective electrolyte accessibility via short ion-transport pathways; (III) Ni3S2 and NiCo2S4 are favorable faradaic electrode materials, whose combination can provide numerous electroactive sites for the faradaic redox reactions, showing a superior capacity. In short, these favorable features will eventually lead to enormous enhancement of electrochemical performance in energy storage.
4. Conclusions
In summary, we have successfully synthesized a unique NiCo2S4@Ni3S2 core–shell NTAs supported on Ni foam through a facile and preferable method with boosted electrochemical performance for supercapacitors. In contrast to bare NiCo2S4 electrodes and other electrode materials, the obtained hierarchical NiCo2S4@Ni3S2 core–shell NTAs electrode exhibits a higher specific capacity and outstanding rate capacity due to its homogeneous core–shell nanotube structure and the faradaic characteristics of NiCo2S4 and Ni3S2. Moreover, the ASC device assembled by taking NiCo2S4@Ni3S2 NTAs as the positive electrode and rGO aerogel as the negative electrode can achieve a mass specific capacity of 163.15 C g−1 at 0.5 A g−1 and a maximum energy density of 32.75 W h kg−1 at a power density of 361 W kg−1, as well as high cycle stability (77.5% retention at 2 A g−1 after 5000 cycles). Even at the highest power density of 2.9 kW kg−1, the NiCo2S4@Ni3S2//rGO device still remains at 25.50 W h kg−1. Therefore, these results not only light up the importance of judiciously designed core–shell nanostructures for achieving enhanced performance, but also demonstrate that NiCo2S4@Ni3S2//rGO holds promising application in ASCs.
Acknowledgements
The authors are grateful for the testing support from the Analysis and Testing Center (Xiamen University).
References
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539–1548 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- R. B. Rakhi, D. Cha, W. Che and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef CAS PubMed.
- Z. Gao, J. Wang, Z. S. Li, W. L. Yang, B. Wang, M. J. Hou, Y. He, Q. Liu, T. Mann, P. P. Yang, M. Zhang and L. H. Liu, Chem. Mater., 2011, 23, 3509–3516 CrossRef CAS.
- S. J. He and W. Chen, Nanoscale, 2015, 7, 6957–6990 RSC.
- M. J. Zhi, C. C. Xiang, J. T. Li, M. Li and N. Q. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- K. Xie, X. T. Qin, X. Z. Wang, Y. N. Wang, H. S. Tao, Q. Wu, L. J. Yang and Z. Hu, Adv. Mater., 2012, 24, 347–352 CrossRef CAS PubMed.
- L. Yu, G. Zhang, C. Yuan and X. W. Lou, Chem. Commun., 2013, 49, 137–139 RSC.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- D. Guo, H. M. Zhang, X. Z. Yu, M. Zhang, P. Zhang, Q. H. Li and T. H. Wang, J. Mater. Chem. A, 2013, 1, 7247–7254 CAS.
- S. L. Xiong, C. Z. Yuan, X. G. Zhang, B. J. Xi and Y. T. Qian, Chem.–Eur. J., 2009, 15, 5320–5326 CrossRef CAS PubMed.
- X. H. Xia, J. P. Tu, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 9319–9325 RSC.
- R. Li, S. L. Wang, J. P. Wang and Z. C. Huang, Phys. Chem. Chem. Phys., 2015, 17, 16434–16442 RSC.
- S. Ratha and C. S. Rout, ACS Appl. Mater. Interfaces, 2013, 5, 11427–11433 CAS.
- X. H. Lu, M. H. Yu, T. Zhai, G. M. Wang, S. L. Xie, T. Y. Liu, C. L. Liang, Y. X. Tong and Y. Li, Nano Lett., 2013, 13, 2628–2633 CrossRef CAS PubMed.
- X. H. Lu, T. Y. Liu, T. Zhai, G. M. Wang, M. H. Yu, S. L. Xie, Y. C. Ling, C. L. Liang, Y. X. Tong and Y. Li, Adv. Energy Mater., 2014, 4, 1300994 CrossRef.
- C. H. An, Y. J. Wang, Y. P. Wang, G. Liu, L. Li, F. Y. Qiu, Y. N. Xu, L. F. Jiao and H. T. Yuan, RSC Adv., 2013, 3, 4628–4633 RSC.
- X. G. Wang, W. Li, D. H. Xiong and L. F. Liu, J. Mater. Chem. A, 2016, 4, 5639–5646 CAS.
- C. C. Hu, J. C. Chen and K. H. Chang, J. Power Sources, 2013, 221, 128–133 CrossRef CAS.
- S. L. Wang, Z. C. Huang, R. Li, X. Zheng, F. X. Lu and T. B. He, Electrochim. Acta, 2016, 204, 160–168 CrossRef CAS.
- Z. L. Wang, R. Guo, G. R. Li, H. L. Lu, Z. Q. Liu, F. M. Xiao, M. Q. Zhang and Y. X. Tong, J. Mater. Chem. A, 2012, 22, 2401–2404 RSC.
- Z. L. Wang, X. J. He, S. H. Ye, Y. X. Tong and G. R. Li, ACS Appl. Mater. Interfaces, 2014, 6, 642–647 CAS.
- H. Yi, H. W. Wang, Y. T. Jing, T. Q. Peng, Y. R. Wang, J. Guo, Q. L. He, Z. H. Guo and X. F. Wang, J. Mater. Chem. A, 2015, 3, 19545–19555 CAS.
- H. C. Gao, F. Xiao, C. B. Ching and H. W. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 2801–2810 CAS.
- X. C. Ren, C. L. Guo, L. Q. Xu, T. T. Li, L. F. Hou and Y. H. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 19930–19940 CAS.
- H. H. Huo, Y. Q. Zhao and C. L. Xu, J. Mater. Chem. A, 2014, 2, 15111–15117 CAS.
- W. T. Wei, L. W. Mi, Y. Gao, Z. Zheng, W. H. Chen and X. X. Guan, Chem. Mater., 2014, 26, 3418–3426 CrossRef CAS.
- R. B. Rakhi, N. A. Alhebshi, D. H. Anjum and H. N. Alshareef, J. Mater. Chem. A, 2014, 2, 16190–16198 CAS.
- L. Zhang, L. Zhou, H. B. Wu, R. Xu and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 7267–7270 CrossRef CAS PubMed.
- W. Kong, C. C. Lu, W. Zhang, J. Pu and Z. H. Wang, J. Mater. Chem. A, 2015, 3, 12452–12460 CAS.
- H. C. Chen, J. J. Jiang, L. Zhang, H. Z. Wan, T. Qi and D. D. Xia, Nanoscale, 2013, 5, 8879–8883 RSC.
- J. Pu, F. L. Cui, S. B. Chu, T. T. Wang, E. H. Sheng and Z. H. Wang, ACS Sustainable Chem. Eng., 2014, 2, 809–815 CrossRef CAS.
- H. Z. Wan, J. J. Jiang, J. W. Yu, K. Xu, L. Miao, L. Zhang, H. C. Chen and Y. J. Ruan, CrystEngComm, 2013, 15, 7649–7651 RSC.
- S. J. Peng, L. L. Li, C. C. Li, H. T. Tan, R. Cai, H. Yu, S. Mhaisalkar, M. Srinivasan, S. Ramakrishna and Q. Y. Yan, Chem. Commun., 2013, 49, 10178–10180 RSC.
- Z. B. Wu, X. L. Pu, X. B. Ji, Y. R. Zhu, M. J. Jing, Q. Y. Chen and F. P. Jiao, Electrochim. Acta, 2015, 174, 238–245 CrossRef CAS.
- W. B. Fu, C. H. Zhao, W. H. Han, Y. Liu, H. Zhao, Y. F. Ma and E. Q. Xie, J. Mater. Chem. A, 2015, 3, 10492–10497 CAS.
- H. Z. Wan, J. Liu, Y. L. Ruan, L. Lv, L. Peng, X. Ji, L. Miao and J. J. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 15840–15847 CAS.
- C. Li and G. Q. Shi, Nanoscale, 2012, 4, 5549–5563 RSC.
- S. B. Ye, J. C. Feng and P. Y. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 7122–7129 CAS.
- S. M. Jung, D. L. Mafra, C. T. Lin, H. Y. Jung and J. Kong, Nanoscale, 2015, 7, 4386–4393 RSC.
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed.
- Q. X. Chu, W. Wang, X. F. Wang, B. Yang, X. Y. Liu and J. H. Chen, J. Power Sources, 2015, 276, 19–25 CrossRef CAS.
- W. Hu, R. Q. Chen, W. Xie, L. Zou, N. Qin and D. H. Bao, ACS Appl. Mater. Interfaces, 2014, 6, 19318–19326 CAS.
- R. Li, S. L. Wang, Z. C. Huang, F. X. Lu and T. B. He, J. Power Sources, 2016, 312, 156–164 CrossRef CAS.
- S. W. Chou and J. Y. Lin, J. Electrochem. Soc., 2013, 160, D178–D182 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- H. C. Chen, J. J. Jiang, L. Zhang, D. D. Xia, Y. D. Zhao, D. Q. Guo, T. Qi and H. Z. Wan, J. Power Sources, 2014, 254, 249–257 CrossRef CAS.
- D. P. Cai, D. D. Wang, C. X. Wang, B. Liu, L. L. Wang, Y. Liu, Q. H. Li and T. H. Wang, Electrochim. Acta, 2015, 151, 35–41 CrossRef CAS.
- J. P. Wang, S. L. Wang, Z. C. Huang and Y. M. Yu, J. Mater. Chem. A, 2014, 2, 17595–17601 CAS.
- J. Y. Zheng, X. Wang, W. Li, Z. W. Cao, H. Wang, C. Zhang, W. G. Song, Y. Ma and J. N. Yao, CrystEngComm, 2012, 14, 7616–7620 RSC.
- T. Zhu, H. B. Wu, Y. B. Wang, R. Xu and X. W. Lou, Adv. Energy Mater., 2012, 2, 1497–1502 CrossRef CAS.
- K. B. Xu, Q. L. Ren, Q. Liu, W. Y. Li, R. J. Zou and J. Q. Hu, RSC Adv., 2015, 5, 44642–44647 RSC.
- W. Li, L. S. Zhang, Q. Wang, Y. Yu, Z. Chen, C. Y. Cao and W. G. Song, J. Mater. Chem., 2012, 22, 15342–15347 RSC.
- L. S. Zhang, W. Li, Z. M. Cui and W. G. Song, J. Phys. Chem. C, 2009, 113, 20594–20598 CAS.
- M. Y. Yu, W. Wang, C. Li, T. Zhai, X. H. Luand and Y. X. Tong, NPG Asia Mater., 2014, 6, e129 CrossRef CAS.
- M. J. Jing, Y. C. Yang, Y. R. Zhu, H. S. Hou, Z. B. Wu and X. B. Ji, Electrochim. Acta, 2014, 141, 234–240 CrossRef CAS.
- Q. X. Chu, W. Wang, X. F. Wang, B. Yang, X. Y. Liu and J. H. Chen, J. Power Sources, 2015, 276, 16–25 CrossRef.
- X. D. Li, Y. Feng, M. C. Li, W. Li, H. Wei and D. D. Song, Adv. Funct. Mater., 2015, 25, 6858–6866 CrossRef CAS.
- X. D. Li, W. Li, M. C. Li, P. Cui, D. H. Chen, T. Gengenbach, L. H. Chu, H. Y. Liu and G. S. Song, J. Mater. Chem. A, 2015, 3, 2762–2769 CAS.
- Y. R. Zhu, X. B. Ji, Z. B. Wu and Y. Liu, Electrochim. Acta, 2015, 186, 562–571 CrossRef CAS.
- Y. Zhu, Z. Wu, M. Jing, X. Yang, W. Song and X. Ji, J. Power Sources, 2015, 273, 584–590 CrossRef CAS.
- H. Yi, H. Wang, Y. T. Jing, T. Q. Peng and X. F. Wang, J. Power Sources, 2015, 285, 281–290 CrossRef CAS.
- D. Cheng, Y. F. Yang, J. L. Xie, C. J. Fang, G. Q. Zhang and J. Xiong, J. Mater. Chem. A, 2015, 3, 14348–14357 CAS.
- L. F. Shen, J. Wang, G. Y. Xu, H. S. Li, H. Dou and X. G. Zhang, Adv. Energy Mater., 2015, 5, 1400977 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21284k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.