
Water stability of self-assembled peptide nanostructures for sequential formation of two-dimensional interstitial patterns on layered materials†
Abstract
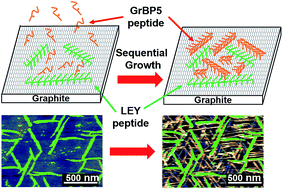
Developing various morphologies and a stable nanostructure of self-assembled peptides on a two-dimensional substrate has played a key role for bioelectronics and biomedical applications. Here, the adsorption and self-assembled characters of two artificial peptides with opposite charges are investigated on graphite and MoS2 surfaces. The ex situ atomic force microscopy (AFM) results show that their morphologies, ordering and stabilities on graphite and MoS2 surfaces are different. The negatively charged peptides self-assembled into an ordered nanostructure on graphite surfaces with six-fold symmetry in a wide range of peptide concentrations. On MoS2 surfaces, the peptide shows a morphology change from randomly orientated nanowires to ordered aligned nanowires as the peptide concentration decreases. On the other hand, the positively charged peptides formed a disordered structure such as wavy structures or aggregates on both substrates. The affinity constants of both peptides on graphite and MoS2 were estimated using concentration-dependence experiments. The stability against water soaking was also examined for both peptides on graphite and MoS2. We found that negatively charged peptides have high affinity constants and stability on both substrates. The results suggest that the stability of self-assembled peptides could be determined by their affinity constants on the substrates. Finally, by using the negatively charged peptides as a stable molecular template, we have demonstrated a sequential self-assembly of two different peptides on a graphite surface. The peptides self-assembled first on the surface show an ability to maintain their nanostructures, and guide the self-assembly of secondary self-assembled peptides. Both of the self-assembled peptides show the same orientations and ordered structures on a graphite surface. These results will open a new door for the development of biosensors with multiple biological probes on a functional surface such as graphite or MoS2.
 Please wait while we load your content...
Please wait while we load your content...

