
Development of biocompatible hydroxyapatite–poly(ethylene glycol) core–shell nanoparticles as an improved drug carrier: structural and electrical characterizations†
Abstract
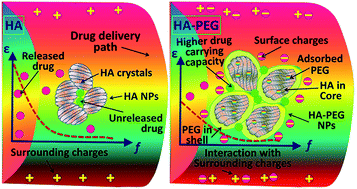
Hydroxyapatite–5 wt% poly(ethylene glycol) (HA–PEG) core–shell composite nanoparticles (NPs) were synthesized using a coprecipitation technique. For the first time, the NPs are characterized for potential drug delivery applications using structural, electrical and in vitro kinetic studies. Phase quantification and the crystal structures of the NPs were analyzed using X-ray diffraction and Fourier transform infrared spectroscopy and the morphology was determined using scanning electron and transmission electron microscopies. Dielectric spectroscopy was used to analyze the polarization behaviour of the HA and HA–PEG core–shell NPs as potential drug carriers by applying an oscillating (100 Hz to 2.5 MHz) electric field. The increased intra-particle interfacial interactions in the HA–PEG NPs confirmed the significant enhancement in interfacial or space charge polarization owing to the reduction in mobility and accumulation of charge-carriers at the interfaces. Thus, HA–PEG showed better aceclofenac drug releasing properties than pristine HA NPs. An in vitro cell study confirmed that the HA and HA–PEG core–shell nanocarriers showed excellent biocompatibility on human dermis fibroblast (HDF) cells. The interaction within the HA–PEG core–shell was stronger than with pristine HA and the biodegradable PEG from the shell-layer neutralized the composite-surroundings path. Hence, it would reduce the direct interaction of aceclofenac drug with the surrounding biomolecules of the delivery paths and enhance the ability for carrying the drug precisely to the target organs.
 Please wait while we load your content...
Please wait while we load your content...

