
Hierarchically porous biomorphic polymer derived C–SiOC ceramics†
Abstract
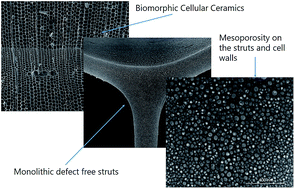
Pinewood derived carbon templates were infiltrated with preceramic polymers and pyrolyzed in inert atmosphere to fabricate hierarchically porous biomorphic silicon oxycarbide amorphous ceramics with ∼80% porosity. Elemental mapping of the ceramics indicated the formation of silicon oxycarbide coating on the carbonaceous skeleton. The pore channels exhibited biomorphic macroporosity, and the channel walls had multimodal mesoporosity as evidenced by N2 adsorption isotherms. Compressive strength of the porous monoliths increased from 0.5 to 4.5 MPa as the pyrolysis temperature was changed from 900 to 1100 °C. The excellent porosity, surface area, and high temperature stability of these materials can be exploited in adsorption, filtration, and catalytic applications.
 Please wait while we load your content...
Please wait while we load your content...

