
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of residual chloride content in ionic liquids using LA-ICP-MS
Maximilian
Bonta
 a,
Thomas
Anderl
a,
Alice
Cognigni
b,
Mahtab
Hejazifar
b,
Katharina
Bica
b and
Andreas
Limbeck
*a
a,
Thomas
Anderl
a,
Alice
Cognigni
b,
Mahtab
Hejazifar
b,
Katharina
Bica
b and
Andreas
Limbeck
*a
aTU Wien, Institute of Chemical Technologies and Analytics, Getreidemarkt 9/164-IAC, 1060 Vienna, Austria. E-mail: andreas.limbeck@tuwien.ac.at
bTU Wien, Institute of Applied Synthetic Chemistry, Vienna, Austria
First published on 14th September 2016
Abstract
Nowadays, ionic liquids (ILs) are used in a wide range of applications. Their exceptional chemical and physical properties, sustainability of use and the possibility of easy recycling are attractive aspects promoting the acceptance of ILs also for commercial applications. While synthesis is in most cases simple and straightforward, purification of the reaction products might pose a number of problems. Due to the major influence of inorganic contaminations from the synthesis process, thorough monitoring of impurities is required. However, the unusual properties of ILs create some problems for conventional chemical analysis. In this work, a dried droplet approach with subsequent LA-ICP-MS sampling will be used for the analysis of chloride in ILs – a by-product from the synthesis procedure. Dried droplet application onto pre-cut filter paper disks allows the analysis of hydrophilic as well as hydrophobic ILs with calibration from dried aqueous standards for signal quantification. The approach is applied on two types of alkylimidazolium ILs, underlining the versatility of the sample preparation and measurement approach. Compared to commonly used analysis techniques for chloride in ILs, the presented approach is simple, fast, and does not require harmful reagents. Different internal standardization strategies were investigated during this study. With a reproducibility of below 2% relative standard deviation and limits of detection of 0.09 mg g−1 for chloride in ILs, the presented approach was showed to be fit for the purpose of routine analyses and reaction monitoring. If necessary, the approach can be extended to other analytes of interest in the field of the synthetic chemistry of ILs.
Introduction
Due to their exceptional chemical and physical properties, ionic liquids (ILs) are increasingly used as a substitute for conventional organic solvents.1 Intrinsic ionic conductivity, good electrochemical properties, and missing volatility are only some of the exceptional features offered by these compounds. This makes them an effective, economic, and sustainable alternative for commonly used solvents. Fields of application are for example chemical synthesis or energy storage. Nowadays, ILs do not only have an application in science, but also have commercial uses.2Alkylimidazolium based ILs are in most cases prepared by alkylation of 1-methylimidazole with an alkyl halide.3–5 The organic salts can then be used for subsequent metathesis reactions with a metal salt e.g., NaBF4, Li(N(Tf)2) or Brønsted acid (e.g., HPF6). Even if this reaction is simple and fast, halide impurities in the synthesized ILs are a considerable issue, especially in the case of tetrafluoroborate ILs.6 Generally, the ease of purification is directly related to the hydrophobicity of the ILs. Excess washing steps with water can be applied to hydrophobic ILs to remove inorganic impurities, whereas the number of possible washing steps is limited for hydrophilic ILs. The remaining chloride content strongly influences the physical properties, such as viscosity, density, or melting point, and alarmingly large discrepancies have been reported in literature for tetrafluoroborate-based ILs.6,7 Additionally, the chloride content can hamper the activity of chemical reactions being carried out in ILs as solvent, due to poisoning or inactivation of catalysts.8,9 While not all applications may require highly purified materials, a precise information on impurities in the synthesized ionic liquid is always required – particularly for ionic liquids that have been prepared via classical anion exchange reactions.10 Reliable determination of the residual chloride content is not a straightforward task due to the high viscosity of the ILs which makes handling and use with conventional sample introduction methods complicated or often impossible. Chemical synthesis laboratories mostly use titrimetric methods such as the Volhard method, or other wet chemical methods; those are often inaccurate, insensitive, and often require the use of harmful and expensive reagents such as AgNO3.11,12 Moreover, results can be questionable in case of hydrophobic ionic liquids, as these methods typically rely on aqueous media.13 Optimized detection methods such as chloride sensors,6 ion chromatography14–16 X-ray fluorescence,17 capillary electrophoresis,18 cathodic stripping voltammetry,19 and even inductively coupled plasma mass spectrometry (ICP-MS)20 have been reported to be employed for residual chloride determination. These methods offer higher sensitivity and accuracy compared to classical wet chemical methods; however, they lack in ease of use and often require sophisticated sample preparation.
An alternative approach for analysis of challenging liquids is the use of dried droplets combined with solid sampling techniques. A liquid sample solution is applied to a carrier material and the solvent is allowed to evaporate. The remaining dried sample spot is then used for chemical analysis; analysis is typically carried out using laser ablation-ICP-MS or electrothermal vaporization atomic absorption spectroscopy (ETAAS).21 When performing laser ablation ICP-MS, a focused laser is fired on a sample leading to the generation of an aerosol which is then transferred and analyzed using an ICP-MS device, providing excellent figures of merit for elemental analysis. Laser ablation ICP-MS has been used for the direct analysis of solid samples already for a few decades and has a wide application range in the geosciences and the life sciences.22–24 It has also been reported to be applied for the sampling of dried liquids on different substrates to circumvent sample digestion of highly matrix loaded samples, for the eased handling of hazardous substances, or for sample pre-concentration.25–27 Nischkauer et al. recently proposed an approach where liquid samples are applied to precut filter pieces, offering the possibility of controlled sample application almost independent of sample viscosity or surface tension.28 It has been shown that aqueous calibration is feasible for signal quantification for samples with heavy matrix load and even for dry samples such as biological tissues.29 This reduces the amount of sample preparation to a minimum and enables fast, reliable, and easy to perform analyses. Moreover, this technique is ideally suited for the analysis of ionic liquids due to their negligible vapor pressure, as a number of solvents (even non-water soluble ones) can be applied for the preparation of the dried ionic liquid droplet, thus allowing to overcome issues with the analysis of hydrophobic ILs due to their limited solubility in aqueous media.
In the presented work, the analysis of the residual chloride content using dried droplets and LA-ICP-MS is demonstrated. Optimization of the experimental procedure enabled the application of aqueous standards for the analysis of ionic liquids. The chloride concentration of two different hydrophobic ionic liquids ([C4mim]N(Tf)2 and [C4mim]BF4) during a series of four washing steps is monitored. The structures of the two investigated ionic liquids are presented in Fig. 1. Application on these ionic liquids demonstrates the suitability of the LA-ICP-MS method for easy, fast, and accurate chloride determination.
 | ||
| Fig. 1 Hydrophobic ionic liquids used in this study. | ||
Methods
Chemicals
Commercially available reagents and solvents were used as received from Sigma Aldrich unless otherwise specified. N-Methylimidazole was distilled from KOH before use. 1-Butyl-3-methylimidazolium chloride was synthesized according to standard methodologies via alkylation of N-methylimidazole with butyl chloride to afford the corresponding imidazolium halide and repeatedly crystallized from EtOAc/CH3CN in order to obtain colorless crystals.30High purity water dispensed from a Barnstead EASYPURE II water system (ThermoFisher Scientific, Marietta, OH) was used for all analytical sample preparation steps. 65% (v/v) nitric acid (p.a.) was purchased from Merck (Darmstadt, Germany).
Preparation of ionic liquids
The ILs were synthesized according to classical anion metathesis procedures available in literature without particular treatment to remove chloride other than stated in the protocols.1-Butyl-3-methylimidazolium bistriflimide [C4mim]N(Tf)2: a solution of LiN(Tf)2 (15.79 g, 55 mmol) in 50 ml water was added dropwise to [C4mim]Cl (8.733 g, 50 mmol) dissolved in 50 ml water. The reaction mixture was stirred for 1 h at room temperature and immediately formed a biphasic system. The reaction mixture was then extracted 4 times with 25 ml CH2Cl2. The organic phases were combined, and a 2 ml sample was taken and evaporated (washing step 0). The remaining organic phase was extracted 3 times with 10 ml H2O each. After every extraction step, a 2 ml sample was taken from the organic layer and evaporated (washing step 1, 2, 3). Finally, the remaining organic phase was dried over Na2SO4 and filtered. The solvent was evaporated, and remaining solvent traces were removed under high vacuum (0.01 mbar) with stirring at room temperature for 24 h to yield 16.916 g [C4mim]N(Tf)2 as colourless liquid. Likewise, all aliquots samples after each washing step (approx. 0.35 g each) were evaporated and dried under high vacuum (0.01 mbar) for 2 h. The overall yield was 87.7%. Analytical data was in accordance with literature.
1-Butyl-3-methylimidazolium tetrafluoroborate [C4mim]BF4:[C4mim]Cl (8.733 g, 50 mmol) and sodium tetrafluoroborate (6.00 g, 55 mmol) suspended in 100 ml anhydrous dichloromethane and stirred under argon atmosphere at room temperature for 24 h. The suspension was filtered over a small batch of celite. A 2 ml sample was taken from the clear organic phase and evaporated (washing step 0). The remaining organic phase was extracted 3 times with 10 ml H2O each. After every extraction step, a 2 ml sample was taken from the organic layer and evaporated (washing step 1, 2, 3). Finally, the remaining organic phase was dried over Na2SO4 and filtered. The solvent was evaporated, and remaining solvent traces were removed under high vacuum (0.01 mbar) with stirring at room temperature for 24 h to yield 5.999 g [C4mim]BF4 as colourless liquid. Likewise, all small samples taken during the washing steps (each approx. 0.12 g) were evaporated and dried under high vacuum (0.01 mbar) for 2 h. The overall yield was 52.3%. Analytical data was in accordance with literature.
Instrumentation
All analytical experiments were performed using a NWR213 laser ablation instrument (ESI, Fremont, CA) coupled to an iCAP Q quadrupole ICP-MS device (ThermoFisher Scientific, Bremen, Germany). Performance of the instrumentation was monitored on a daily basis using NIST612 trace elements in glass standard reference material (National Institute of Standards and Technology, Gaithersburg, MD) using the sensitivity for 115In signal. Typical parameters of the LA-ICP-MS measurements are presented in Table 1.| Laser ablation | ICP-MS | ||
|---|---|---|---|
| Laser wavelength | 213 nm | Plasma power | 1500 W |
| Laser energy | 14.6 mJ | Cool gas flow | 13.0 l min−1 |
| Laser beam diameter | 200 μm | Aux gas flow | 0.8 l min−1 |
| Laser repetition rate | 20 Hz | Scanned isotopes | 35Cl, 79Br, 81Br, 113In |
| Scan speed | 100 μm s−1 | Scan mode | Peak hopping |
| He gas flow | 750 ml min−1 | Mass resolution | 300 m/Δm |
| Ar make-up flow | 800 ml min−1 | Quadrupole dwell time | 10 ms |
The LA instrumentation was equipped with a fast washout ablation cell provided by the manufacturer of the instrument. Ablation of the sample material was performed under a constant stream of helium, which was mixed with argon make-up gas upon introduction into the ICP-MS device.
Sample preparation and LA-ICP-MS measurements
Due to high viscosity and delicate handling properties of the ILs, all samples and standards were weighed to increase accuracy of the determinations. Thus, all results are given in mg g−1. Aqueous chloride standard solutions were prepared from potassium chloride dissolved in high purity water. Concentration levels of 0.5, 1.0, 2.0, 5.0, and 10.0 mg g−1 chloride were prepared from a stock solution containing 100.0 mg g−1 chloride. Additionally, a blank solution without added chloride was prepared. For evaluations on the suitability of an internal standardization approach, indium – a commonly used internal standard in ICP-MS – was investigated. It was added to all solutions at a final concentration of 10.0 μg g−1 in the form of an aqueous indium solution (indium for ICP, 1000 g l−1). Bromine was the alternative candidate to be used as internal standard. It was added to all solutions in the form of potassium bromide to obtain a final bromide concentration of 1.0 mg g−1.10 μl of the prepared standards were applied to the pre-cut filter pieces and allowed to let dry at room temperature. Due to incomplete solubility of [C4mim]N(Tf)2 in water, a two-step procedure for preparation of samples and spiked samples had to be employed. In the first step, 10 μl of aqueous solutions of chloride (1.0, 5.0, 10.0 mg g−1, and a blank) were applied to pre-cut filters and allowed to dry completely at room temperature. Spike concentrations were chosen to be in the expected concentration range of chloride in the samples. In a second step, all ionic liquids were diluted in ethanol containing 2.0 mg g−1 bromide (from KBr) by a factor of 2.0 (m m−1). 5 μl of thereby diluted ionic liquid were applied to the previously prepared filter pieces. The solvent was allowed to evaporate at room temperature before proceeding with the measurements. Four replicates of each standard and sample/spike were prepared. This approach allowed a very reproducible formation of dried IL-droplets on the filters, as well as high flexibility regarding the analysed IL types.
All prepared samples were measured with radial line-scans across the diameter of the filter pieces, as reported previously. Laser parameters were kept constant throughout all experiments and were chosen to completely ablate the filter material exposed by the laser beam. During selection of the parameters, care has been taken to ensure smooth ablation of all samples in order to obtain a constant signal-over-time profile during LA-ICP-MS analysis. In this context, the term ‘smooth ablation’ describes the fact that only the sample directly irradiated by the laser beam is ablated; no ejection of larger particles from surrounding areas occurs. For data evaluation, the signals measured across the whole scans were integrated; background signals were collected prior to every sample measurement and subtracted from the measured intensities. Average values of the four replicates were calculated; standard deviations of the calculated means were used for uncertainty estimations. 37Cl was not considered for analysis due to a possible spectral interference by 36Ar1H.
Results
Analyte distribution across pre-cut filter pieces
Drying of liquids on filters is known to be influenced by the so-called coffee stain effect. This phenomenon describes chromatographic effects occurring during drying of a droplet.31 A result of the coffee stain effect is the uneven analyte distribution on the filter with its maximum towards the edges of the filter piece. However, in an ideal case, this distribution is symmetric around the center of the filter circle. To obtain reliable and consistent results using the radial line-scan method, it is imperative that this prerequisite is met. Especially in the case of ionic liquids which do not dry and just get soaked up by the filter material, symmetric analyte distribution is a critical aspect. In Fig. 2, typical signal-over-time profiles of scans across an aqueous standard sample (1.0 mg g−1 chloride) and a non-spiked ionic liquid sample are presented. Additionally, the signal of a blank sample is shown, indicating the low background signals originating from blank filters. Replicate measurements show no significant difference from the depicted profiles.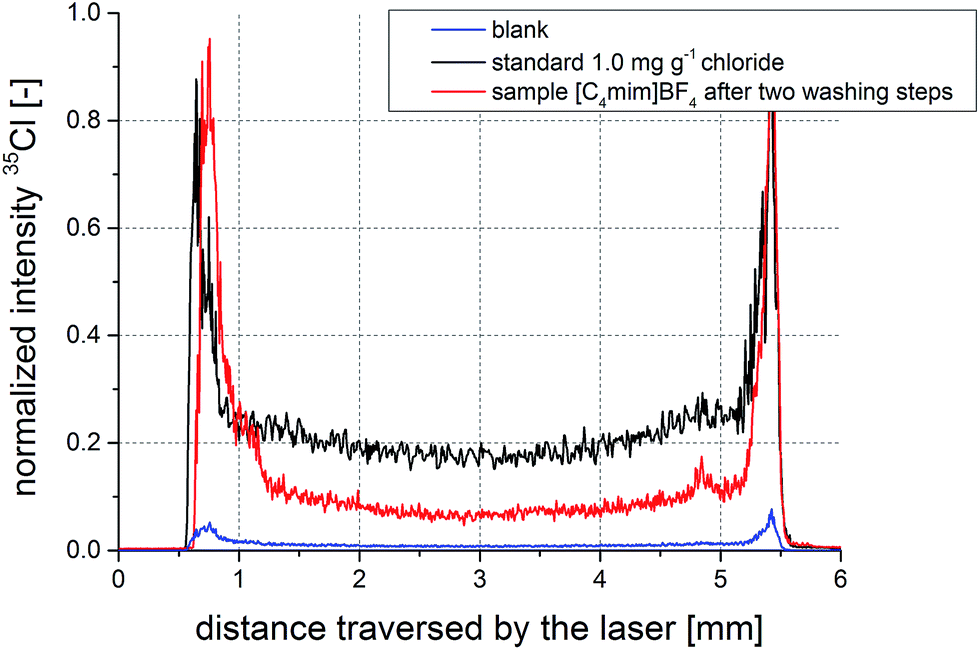 | ||
| Fig. 2 Signal-over-time profiles from scans across filters with aqueous standard (black), an ionic liquid sample applied after dissolution in ethanol (red), and a blank solution (blue). | ||
The signal profiles are symmetric and indicate the feasibility of the radial line-scan approach for IL measurements. Additionally, the distributions of chloride appear to be very similar for aqueous standard and ionic liquid, not indicating any excessive analyte accumulation at the border regions of the filter. Ethanol does not seem to have a negative effect on the proper analyte distribution. Sample and standard signals are well above background level, showing suitability of the proposed method for the investigated concentration ranges of the samples.
Quantification of chloride contents in ionic liquids
Compared to aqueous dried droplet standards, the ablation behavior as well as the aerosol transport and the analyte ionization may differ significantly for ionic liquid samples. Such matrix-related differences in material ablation can in the case of ILs especially be contributed to the fact that an IL will not completely dry on a filter. After evaporation of the solvent, the highly viscous IL will remain, being soaked up by the filter material. This may result in variations in laser–material interaction, as well as transport efficiency of the generated aerosol. Such variations can be compensated using standard addition, where all measured samples basically exhibit the same matrix composition. Thus, the standard addition method represents the most accurate quantification strategy with regards to matrix variations. In this experiment, spike levels of all ILs were 1.0, 5.0, and 10.0 mg g−1 chloride. Integrated signals for 35Cl ranged between 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cts and 7000000 cts and were significantly different from the gas-blank in all cases. Reproducibility of the measurements was always below 10.0% relative standard deviation (RSD) for four replicates of sample preparation. This is a satisfactory value as the variation is besides measurement uncertainty also including differences in sample preparation. Slopes of the regression functions used for data evaluation using the standard addition method were never below 0.99; the absolute values of the slopes for the two investigated ionic liquids differed significantly. Concentrations between 0.62 and 7.9 mg g−1 chloride for different numbers of washing steps were found in both ILs, indicating differences in the chloride content at increasing number of washing steps.
000 cts and 7000000 cts and were significantly different from the gas-blank in all cases. Reproducibility of the measurements was always below 10.0% relative standard deviation (RSD) for four replicates of sample preparation. This is a satisfactory value as the variation is besides measurement uncertainty also including differences in sample preparation. Slopes of the regression functions used for data evaluation using the standard addition method were never below 0.99; the absolute values of the slopes for the two investigated ionic liquids differed significantly. Concentrations between 0.62 and 7.9 mg g−1 chloride for different numbers of washing steps were found in both ILs, indicating differences in the chloride content at increasing number of washing steps.
Internal standardization and calibration with aqueous solutions
As the workload of standard addition experiments is significantly higher than for external calibrations, it was desirable to develop a feasible method for external calibration of the chloride contents in the ionic liquids. Thus, aqueous chloride standards were used to determine a calibration for signal quantification. However, as a significant matrix difference between dried droplets from ionic liquids and aqueous standards was expected, the use of a suitable internal standard needed to be evaluated. Measurement of aqueous standards (four replicates of sample preparation for each standard) of chloride yielded signal intensities between 500000 cts and 12000000 cts (0.5–10.0 mg g−1) compared to 20000 cts for the blank (10 μl standard solution per filter). A calibration from raw 35Cl signals is shown in Fig. 3. While the calibration function exhibits a good correlation coefficient of 0.9994, reproducibility of replicate standard preparations of up to 10% is not perfect for accurate calibration and therefore requires improvement. This variation might be explained by inaccuracies during pipetting of the aqueous standards onto the filter paper disks. Fig. 3 also shows a comparison of the calibration function from the aqueous standards with a standard addition experiment from sample [C4mim]N(Tf)2 before the first washing step. The slopes of the regression functions differ significantly, indicating a matrix effect that influences material ablation, transport, and/or analyte ionization. The slope of the standard addition curve is lower than the aqueous calibration, which indicates a weaker detection efficiency of chloride in the ionic liquid samples. This might be contributed to the fact that the aerosol generated by the laser contains a ‘sticky’ fraction: if the ionic liquid is not completely decomposed in the laser generated plasma, parts of the aerosol will consist of remaining IL which can then adhere to tubing of the aerosol transfer line. Thus, an internal standard is required to achieve the goal of signal quantification using an aqueous calibration.
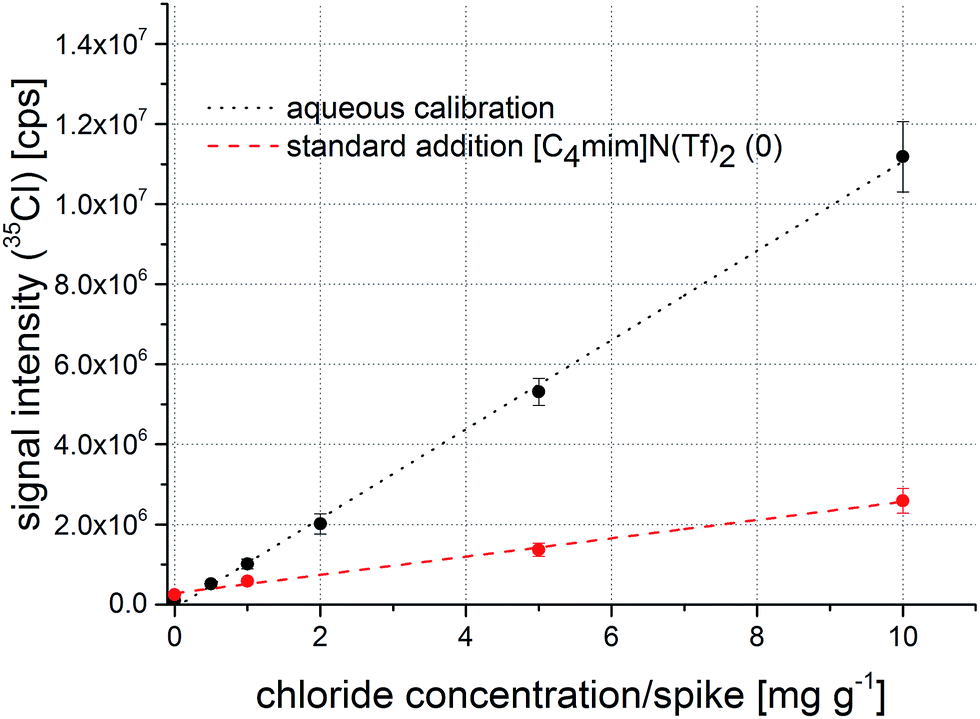 | ||
| Fig. 3 Calibration function obtained from aqueous standard solutions (black) and a regression function from a standard addition experiment (red) without signal normalization to an internal standard. | ||
Firstly, indium was evaluated as a candidate for an internal standard. Again, a good correlation coefficient of 0.9992 for the regression function was calculated. Results show that the reproducibility for replicate sample preparation improves to around 5%, while the slope of the resulting regression function is still significantly different from the standard addition experiment. In analogy to Fig. 3, a comparison of aqueous calibration and standard addition using indium as internal standard is shown in Fig. 4. The absolute values of the intercepts of the regression functions converge compared to the raw 35Cl calibration. Thus, it can be stated that indium helps to minimize the pipetting errors, while it is not capable to correct all existing matrix effects. Possibly, differences in material transport are reduced as both analytes travel together in the aerosol but ionization variances in the ICP are not tackled by indium as internal standard; the large difference in the first ionization potential (chlorine: 1251.2 kJ mol−1, indium: 558.30 kJ mol−1) certainly plays a major role.
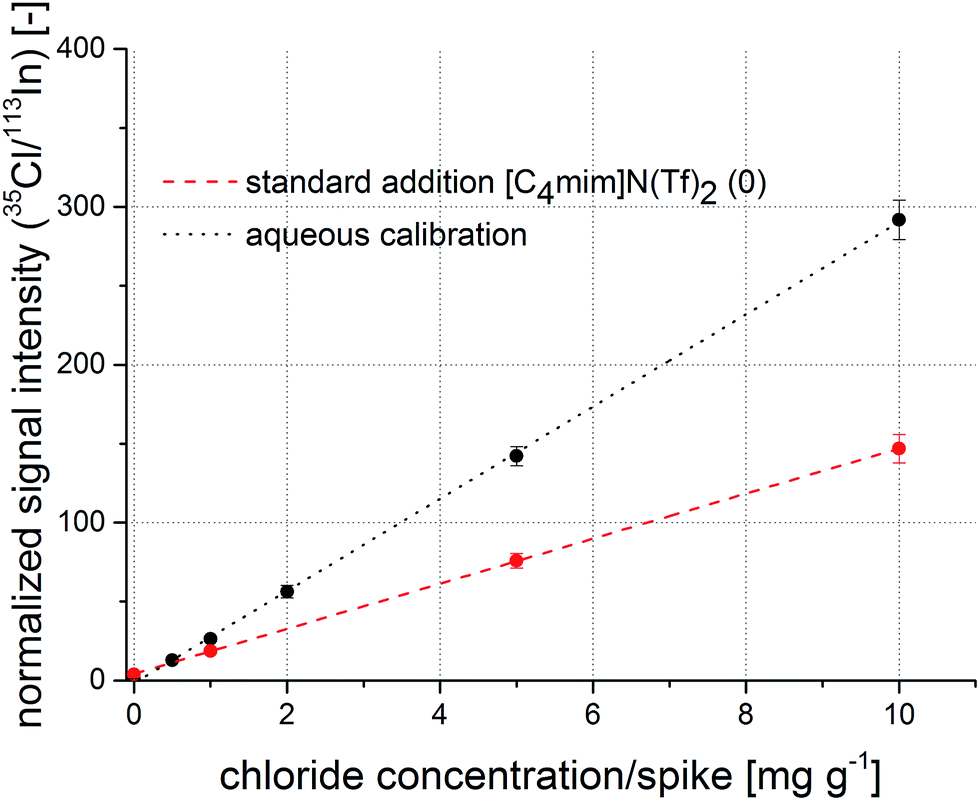 | ||
| Fig. 4 Calibration function obtained from aqueous standard solutions (black) and a regression function from a standard addition experiment (red) with normalization to indium as internal standard. | ||
The second candidate for an internal standard was bromine. Due to a comparable first ionization potential with chlorine (chlorine: 1251.2 kJ mol−1, bromine: 1139.9 kJ mol−1) and similar chemical properties, this element was selected. Normalization of the 35Cl signals to the 79Br signals indicates that reproducibility of replicate measurements is again improved compared to measurements without internal standard. With a maximum of 6% relative standard deviation and a correlation coefficient of 0.9996, these figures are comparable with the indium normalization. Additionally, values for the intercept of the calibration function does not differ significantly from the standard addition method at a significance level of 95%. Measurements of the other ionic liquid indicate similar results. The respective regression functions are presented in Fig. 5. The offset on the y-axis originates from the initial chloride concentration in the unspiked sample. The combination of filter matrix and use of 79Br as internal standard shows to correct previously described ablation, transport, and ionization differences of the analyte in the presence of IL matrix.
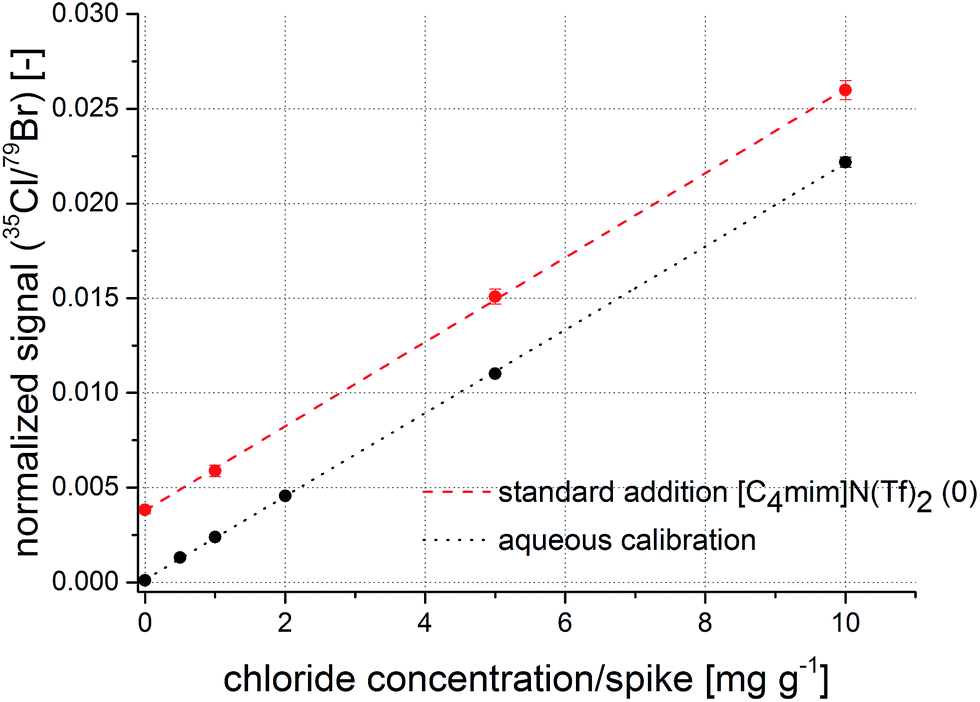 | ||
| Fig. 5 Calibration function obtained from aqueous standard solutions (black) and a regression function from a standard addition experiment (red) with normalization to bromine as internal standard. | ||
Based on the aqueous calibration, a limit of detection of 0.09 mg g−1 and a limit of quantification of 0.16 mg g−1 chloride can be determined. Compared to typical metallic analytes, detection power for chlorine is considerably lower. Investigations on solutions containing transition metals (e.g., Cu, Fe, Zn) performed in the same laboratory and using the same sample volume resulted in typical limits of detection around 0.1 μg g−1. Considering different volumes of sample intake, this value is in good accordance with other studies reported in literature.25 This large difference in the limits of detection can mainly be explained by the high first ionization potential of chlorine and the thereby resulting weak detection efficiency in the ICP-MS compared to metallic analytes investigated in other studies. Using this calibration, signals obtained from the sample measurements were quantified and compared to the results from the standard addition approach. Results of both quantification methods are shown in Table 2.
| Ionic liquid | Washing step | Standard addition | External calibration | ||
|---|---|---|---|---|---|
| Concentration [mg g−1] | Standard deviation | Concentration [mg g−1] | Standard deviation | ||
| [C4mim]N(Tf)2 | 0 | 1.65 | 0.11 | 1.67 | 0.10 |
| 1 | 1.04 | 0.05 | 1.07 | 0.03 | |
| 2 | 0.59 | 0.04 | 0.62 | 0.02 | |
| 3 | 0.62 | 0.05 | 0.65 | 0.03 | |
| [C4mim]BF4 | 0 | 7.88 | 0.24 | 7.88 | 0.21 |
| 1 | 4.22 | 0.24 | 4.24 | 0.22 | |
| 2 | 1.28 | 0.08 | 1.30 | 0.07 | |
| 3 | 0.83 | 0.04 | 0.86 | 0.04 | |
After testing for normal distribution and equal variances, a t-test was employed to validate if the obtained results vary significantly. No indication was found that there is a difference between both obtained concentrations at a 95% level of significance. Additionally using the aqueous calibrations, recoveries were calculated from the analysis of the spiked samples; found spike concentrations were compared to the added amounts of chloride. Values between 94 and 103% were yielded.
Application of chloride quantification using LA-ICP-MS for quality control in IL synthesis
For the determined concentrations from four washout steps, a clear trend can be observed; as expected, a higher number of washing steps lowers the amount of chloride. When comparing the chloride contents in the two ionic liquids, the more hydrophilic IL ([C4mim]BF4) exhibits higher chloride concentrations than [C4mim]N(Tf)2. While in the case of [C4mim]N(Tf)2 the chloride concentration after the first washing step is 1.6 mg g−1, the value for [C4mim]BF4 is almost a factor of 5 higher. However, after four washout steps, the chloride concentrations found in both ILs are comparable. It is important to notice that considerable residual chloride content was present in both ionic liquids after these few washing steps that are often routinely applied during synthesis. Moreover the commonly used “silver nitrate” test has been used to detect halides present in the water layer during the extraction steps might already give false negative results during the synthesis, as the limit of detection might vary for ionic liquids with different water miscibility.6 Eventually, our results can be seen as clear warning and emphasize the importance of careful ionic liquid synthesis and purification in combination with a reliable detection method, since variable residual chloride contents after a few routine washing steps will hamper any future application.Conclusion and outlook
We presented a novel approach for the analysis of residual inorganic contaminants in ionic liquids. Our strategy relies on the low volatility of ILs and is based on a preliminary dilution of the sample with ethanol to simplify further sample handling. After deposition of a sample droplet on a pre-cut filter paper disk, the solvent is smoothly evaporated, and finally the filter disk containing the non-volatile ionic liquid is analyzed using LA-ICP-MS. After thorough optimization, the developed approach enables external calibration using aqueous standard solutions, and thus offers a simplified method for accurate quantification of chloride contaminations in hydrophobic ionic liquids. Compared to existing approaches for chloride determination, the proposed method offers a number of advantages. Important features are selectivity, sensitivity, the low required sample amount, simple quantification and the possibility to analyze hydrophilic as well as hydrophobic ILs. In comparison to the commonly used titrimetric Volhard method, the use of toxic and environmentally harmful reagents is circumvented. Furthermore, selectivity of the wet chemical approach might be influenced by interfering ions, and the time-effort required for the titrimetric analysis is significantly larger. While chromatographic methods showed similar limits of detection for chloride determination,16 elution can be severely influenced by the structural properties of a specific IL, making the method less versatile for routine applications where a large range of different ILs needs to be analyzed.15 Limits of detection are as well comparable with the X-ray based chloride analysis reported by Vander Hoogerstraete et al.17 However, sample preparation is more tedious in this protocol, hampering the suitability for routine application. For cathodic stripping voltammetry, better limits of detection have been reported,19 but larger experimental effort has to be taken into account for this analysis method. Summing up, the presented method for chloride determination in IL samples using LA-ICP-MS shows to be competitive with existing methods regarding the analytical performance. However, sample preparation and sample consumption can be reduced to a minimum. Only environmentally friendly reagents are used during the analysis procedure, making the presented approach a green alternative to currently available approaches. Possible application for multi-element analysis is also a great advantage; besides chloride it is possible to also measure other elemental analytes in the samples, quantification of other elements can be easily implemented into the existing method.References
- M. Freemantle, An introduction to ionic liquids, 2009, http://www.rsc.org/shop/books/2009/9781847551610.asp Search PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- C. M. Gordon, M. J. Muldoon, M. Wagner, C. Hilgers, J. H. Davis and P. Wasserscheid, in Ionic Liquids in Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 7–55, DOI:10.1002/9783527621194.ch2.
- M. Deetlefs and K. R. Seddon, Green Chem., 2003, 5, 181–186 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- K. R. Seddon, A. Stark and M. J. Torres, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS.
- J. Jacquemin, R. Ge, P. Nancarrow, D. W. Rooney, M. F. Costa Gomes, A. A. H. Pádua and C. Hardacre, J. Chem. Eng. Data, 2008, 53, 716–726 CrossRef CAS.
- S. H. Lee, S. H. Ha, S. B. Lee and Y. M. Koo, Biotechnol. Lett., 2006, 28, 1335–1339 CrossRef CAS PubMed.
- A. Stark, M. Ajam, M. Green, H. G. Raubenheimer, A. Ranwell and B. Ondruschka, Adv. Synth. Catal., 2006, 348, 1934–1941 CrossRef CAS.
- A. Stark, P. Behrend, O. Braun, A. Müller, J. Ranke, B. Ondruschka and B. Jastorff, Green Chem., 2008, 10, 1152–1161 RSC.
- P. Wasserscheid, R. Van Hal and A. Bösmann, Green Chem., 2002, 4, 400–404 RSC.
- P. A. Z. Suarez, J. E. L. Dullius, S. Einloft, R. F. De Souza and J. Dupont, Polyhedron, 1996, 15, 1217–1219 CrossRef CAS.
- A. I. Vogel, A textbook of quantitative analysis including instrumental analysis: with diagrams and illus, John Wiley and Sons, New York, 1961 Search PubMed.
- J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef CAS PubMed.
- F. Hao, P. R. Haddad and T. Ruther, Chromatographia, 2008, 67, 495–498 CAS.
- C. Villagrán, M. Deetlefs, W. R. Pitner and C. Hardacre, Anal. Chem., 2004, 76, 2118–2123 CrossRef PubMed.
- T. Vander Hoogerstraete, S. Jamar, S. Wellens and K. Binnemans, Anal. Chem., 2014, 86, 3931–3938 CrossRef CAS PubMed.
- A. Markowska and P. Stepnowski, J. Sep. Sci., 2010, 33, 1991–1996 CrossRef CAS PubMed.
- C. Villagrán, C. E. Banks, C. Hardacre and R. G. Compton, Anal. Chem., 2004, 76, 1998–2003 CrossRef PubMed.
- K. McCamley, N. A. Warner, M. M. Lamoureux, P. J. Scammells and R. D. Singer, Green Chem., 2004, 6, 341–344 RSC.
- L. Rello, A. C. Lapeña, M. Aramendía, M. A. Belarra and M. Resano, Spectrochim. Acta, Part B, 2013, 81, 11–19 CrossRef CAS.
- R. E. Russo, X. Mao, J. J. Gonzalez, V. Zorba and J. Yoo, Anal. Chem., 2013, 85, 6162–6177 CrossRef CAS PubMed.
- D. Günther and B. Hattendorf, TrAC, Trends Anal. Chem., 2005, 24, 255–265 CrossRef.
- A. Limbeck, P. Galler, M. Bonta, G. Bauer, W. Nischkauer and F. Vanhaecke, Anal. Bioanal. Chem., 2015, 407, 6593–6617 CrossRef CAS PubMed.
- M. Aramendía, L. Rello, F. Vanhaecke and M. Resano, Anal. Chem., 2012, 84, 8682–8690 CrossRef PubMed.
- L. Yang, R. E. Sturgeon and Z. Mester, Anal. Chem., 2005, 77, 2971–2977 CrossRef CAS PubMed.
- H.-F. Hsieh, W.-S. Chang, Y.-K. Hsieh and C.-F. Wang, Talanta, 2009, 79, 183–188 CrossRef CAS PubMed.
- W. Nischkauer, F. Vanhaecke, S. Bernacchi, C. Herwig and A. Limbeck, Spectrochim. Acta, Part B, 2014, 101, 123–129 CrossRef CAS PubMed.
- M. Bonta, B. Hegedus and A. Limbeck, Anal. Chim. Acta, 2016, 908, 54–62 CrossRef CAS PubMed.
- J. Dupont, C. S. Consorti, P. A. Z. Suarez, R. F. De Souza, S. L. Fulmer, D. P. Richardson, T. E. Smith and S. Wolff, Org. Synth., 2002, 79, 236–243 CrossRef CAS.
- G. Berteloot, A. Hoang, A. Daerr, H. P. Kavehpour, F. Lequeux and L. Limat, J. Colloid Interface Sci., 2012, 370, 155–161 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |