
Coordination-driven multilayer of phosvitin-polyphenol functional nanofibrous membranes: antioxidant and biomineralization applications for tissue engineering†
Abstract
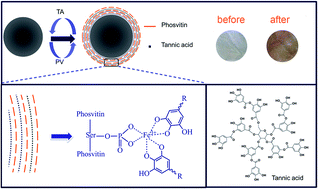
The layer-by-layer (LBL) deposition technique has been widely used to decorate the nanofibers formed from polymer pairs with complementary functional groups. In the current study, an antioxidative coordination-driven multilayer electrospun nanofibrous film was fabricated from tannic acid (TA) and phosvitin (PV) obtained from egg yolk. PV was reported to bond with 95% of yolk iron which could provide chelating sites for TA. The surface morphology of the nanofibrous mats was observed by scanning electron microscopy (SEM). Also the TEM image of the cross-section illustrated a uniform shell formed around the cellulose nanofibers. The deposition of TA and PV was further confirmed by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FT-IR). TA/PV nanofibrous mats showed good 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical-scavenging activity regardless of the outmost component. While for superoxide-scavenging and hydroxyl radical-scavenging activity, the outmost component affected the scavenging capacity of nanofibrous mats. Scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) were combined to characterize the morphology and structure of the deposited mineral phase on the scaffolds after culturing in a simulated body fluid (SBF) for 5 days. From the result, the TA/PV nanofibrous mats were satisfactory for use in bioapplications.
 Please wait while we load your content...
Please wait while we load your content...

