
Degradation of the active species in the catalytic system Pd(OAc)2/NEt3†

Abstract
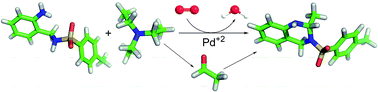
The degradation under ambient humidity and room temperature of Pd(OAc)2(NEt3), which is an efficient catalyst for the aerobic oxidation of alcohols, to diethylamine and acetaldehyde derivatives is disclosed. Evolved diethylamine reacted with Pd(OAc)2 to give Pd(OAc)2(HNEt2), which is in dynamic equilibrium with Pd(OAc)2(HNEt2)2. The evolved acetaldehyde generated during degradation process was trapped with a nucleophile to form 2-methyl-3-tosyl-1,2,3,4-tetrahydroquinazoline. NMR spectroscopy and single crystal X-ray diffraction studies have been used to characterize the reaction products of the reaction system.
 Please wait while we load your content...
Please wait while we load your content...

