
Copper-catalyzed one-pot oxidative amidation of alcohol to amide via C–H activation†
Jiajia Gua,
Zheng Fang*a,
Yuhang Yanga,
Zhao Yangb,
Li Wana,
Xin Lia,
Ping Weia and
Kai Guo *ac
*ac
aCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China. E-mail: guok@njtech.edu.cn; fzcpu@163.com; Fax: +86 25 5813 9935; Tel: +86 25 5813 9926
bSchool of Engineering, China Pharmaceutical University, No. 639 Longmian Avenue, Nanjing 211198, China
cState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China
First published on 14th September 2016
Abstract
A one-pot oxidative amidation of both aliphatic and aromatic alcohols with N-chloramines, prepared in situ from many types of primary and secondary amines, was developed. This cross-coupling reaction integrates alcohol oxidation and amide bond formation, which are usually accomplished separately, into a single operation. And it was green, simple and convenient, which has a wide substrate scope and makes use of cheap, abundant, and easily available reagents. The practical value of this method is highlighted through the synthesis of a high-profile pharmaceutical agent, acetylprocainamide.
The formation of amide bonds is one of the most often used organic reactions due to the extensive presence of this functional group in natural products, pharmaceutical compounds, and synthetic polymers.1 Conventionally, amides are synthesized by the reaction of an amine with carboxylic acid, which needs either a coupling reagent2 or conversion into more reactive derivatives.3 However, these methods have several common shortcomings, such as poor atom-efficiency, use of hazardous reagents, and generation of waste which causes environmental problems. To solve these problems, alternative methods for amide synthesis were developed, such as named reactions like the Schmidt reaction,4 the Beckmann rearrangement,5 and Ugi reaction.6 A more recent approach has considered the use of direct oxidative amidation from aldehydes.7 However, these methods require the use of expensive transition metals as catalyst, and the use of aldehydes can be troublesome due to their inherent reactivity.
From the recent perspective of green chemistry, it remains a great challenge to develop a general and efficient method for the synthesis of amide. The direct oxidative amidation of alcohols can be a potentially attractive method due to the use of cheap, abundant and stable starting materials.8 To date, the oxidative amidation of alcohols is essentially promoted by homogeneous or heterogeneous catalysts, such as Ru- or Rh-based transition metal complexes,9 and Ag- or Au-based supported catalysts.10 Usually, these strategies consist of the oxidation of an alcohol to the corresponding aldehyde that reacts with an amine and then further oxidize the hemiaminal to the desired amide (Scheme 1).
 | ||
| Scheme 1 Oxidative amidation of alcohols with amines. | ||
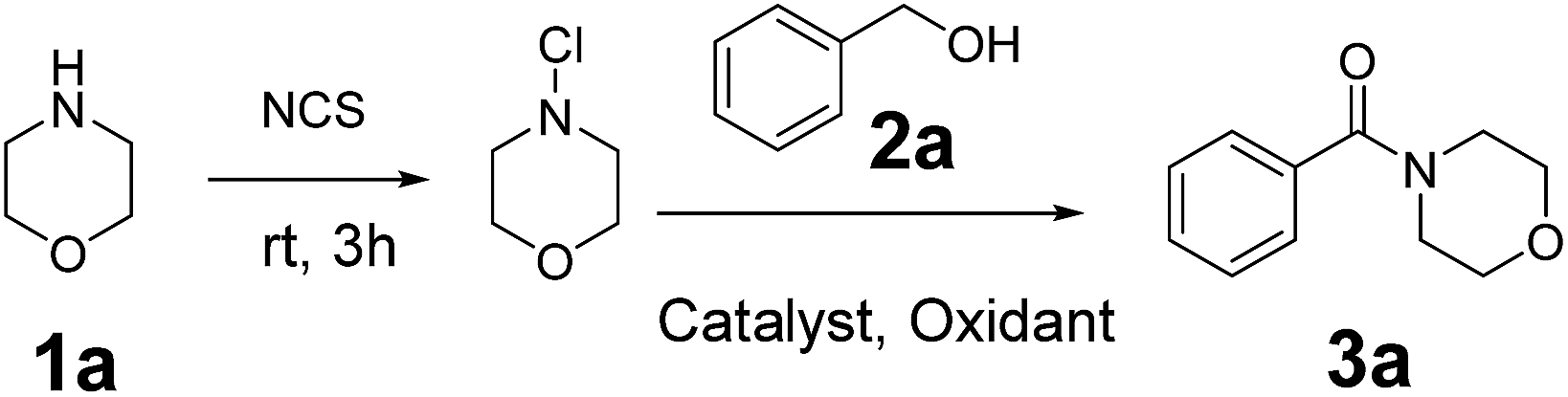
In order to compete with those techniques involving expensive metals, studies on zinc- or copper- or iron-catalyzed oxidative amidation of alcohols have been developed.11–13 In 2013, Wu et al. reported the zinc-catalyzed oxidative amidation of benzyl alcohols (Scheme 2a).11 Similar approach using copper or iron was developed (Scheme 2b).12,13 However, these methods have several drawbacks, such as the formation and stability of the hemiaminal intermediate, the use of expensive transition metal catalysts, and the limited substrate scope. Therefore, it remains a great challenge to develop new methods to synthesis amide. The direct conversion of C–H bonds into C–N bonds appears to be a critical but appealing challenge in organic chemistry. Recently, Wan14 and Wang15 reported the formation of C–N bond via C–H aldehyde bond activation. In 2013, Gaspa et al. used this method to develop the iron-catalyzed amidation of N-chloramines, prepared from amine, which needed the use of 5 equiv. of alcohols to obtain good yields (Scheme 2c).16 However, these methods outlined above shared a common drawback, which was that they showed excellent activity only with benzyl alcohols. Aliphatic alcohols did not perform well in their system. Herein, we report an efficient and green one-pot procedure for the direct amidation of both aliphatic and aromatic alcohols with N-chloramines, which were prepared from amines and without purification, catalyzed by easily accessible copper salts under base-free conditions (Scheme 2d).
 | ||
| Scheme 2 Metal-catalyzed oxidative amidation of alcohols with amines. | ||
At the beginning, morpholine 1a (1 equiv.) was treated with N-chlorosuccinimide (NCS) (1.1 equiv.) in acetonitrile at room temperature for 3 h, and then benzyl alcohol 2a (1.2 equiv.), Cu(OAc)2 (10 mol%) and TBHP (70 wt% in water, 5 equiv.) were added to the reaction mixture under 80 °C to generate the amide product 3a in only 30% yield (Table 1, entry 1). In order to improve the yield of the product, different parameters such as catalyst, oxidant and stoichiometric mole ratio of reactants were examined. Firstly, different catalysts were used instead of Cu(OAc)2 to test the reaction and CuSO4·5H2O performed best among these catalysts with a yield of 62% (Table 1, entries 1–5). Then different oxidants were used to evaluate the reaction. When we used TBPB as an oxidant, the yield of the product was decreased apparently (Table 1, entry 6). And the product obtained only in trace when we used H2O2 or m-CPBA as oxidant (Table 1, entries 7 and 8). In the absence of an oxidant, there was no product observed (Table 1, entry 9). When the amount of catalyst increased to 20 mol%, the yield of product was improved significantly (Table 1, entry 10). And it was unnecessary to continue to increase the amount of catalyst (Table 1, entry 11). The decrease in the amount of TBHP from 5 equiv. to 3 equiv. led to a collapse of the yield (Table 1, entry 12).
|
|||
|---|---|---|---|
| Entry | Catalyst | Oxidant | Yieldb (%) |
| a Reaction conditions: morpholine (1.5 mmol), NCS (1.65 mmol, 1.1 equiv.), in 4 mL acetonitrile, stirring at room temperature for 3 h. To this reaction mixture were added benzyl alcohol (1.8 mmol, 1.2 equiv.), catalyst (10 mol%) and oxidant (7.5 mmol, 5 equiv.), stirring at 80 °C for 24 h.b Isolated yield.c Reaction performed using 20 mol% of CuSO4·5H2O.d Reaction performed using 50 mol% of CuSO4·5H2O.e Reaction performed using 20 mol% of CuSO4·5H2O and 3 equiv. of TBHP. | |||
| 1 | Cu(OAc)2 | TBHP | 30 |
| 2 | Cu(OAc)2·H2O | TBHP | 34 |
| 3 | CuBr | TBHP | 42 |
| 4 | CuI | TBHP | 28 |
| 5 | CuSO4·5H2O | TBHP | 62 |
| 6 | CuSO4·5H2O | TBPB | 48 |
| 7 | CuSO4·5H2O | H2O2 | Trace |
| 8 | CuSO4·5H2O | m-CPBA | Trace |
| 9 | CuSO4·5H2O | — | — |
| 10c | CuSO4·5H2O | TBHP | 92 |
| 11d | CuSO4·5H2O | TBHP | 90 |
| 12e | CuSO4·5H2O | TBHP | 44 |
With optimized reaction conditions in hand, a wide range of commercial available alcohols was used to test the substrate scope of this reaction. And the results were summarized in Table 2. Aromatic alcohols with both electron-rich group, such as CH3 (Table 2, 3b) and OMe (Table 2, 3c), and electron-deficient group, such as NO2 (Table 2, 3d), Cl (Table 2, 3e) and Br (Table 2, 3f), were well tolerated to provide the desired amide products in good to excellent yields. Similarly, heterocyclic alcohols 2g and 2h gave the corresponding amide products in good yields (Table 2, 3g and 3h). Moreover, when aliphatic alcohols, such as n-butanol and n-hexanol, were employed as the substrate, the corresponding amide products were obtained in moderate yield (Table 2, 3i–3l).
| a Reaction conditions: amine (1.5 mmol), NCS (1.65 mmol, 1.1 equiv.), in 4 mL acetonitrile, stirring at room temperature for 3 h. To this reaction mixture were added alcohol (1.8 mmol, 1.2 equiv.), CuSO4·5H2O (20 mol%) and TBHP (7.5 mmol, 5 equiv.), stirring at 80 °C for 24 h. |
|---|
 |
Next, an array of commercially available amines was used to test the substrate scope of this reaction. And the results were shown in Table 3. Disubstituted amines, such as cyclic (Table 3, 3a, 3m–3o) and acyclic amines (Table 3, 3p–3t), showed great activity to obtain the product in good yields. Furthermore, monosubstituted amines were also showing excellent tolerance (Table 3, 3u–3z), even when they were sterically hindered (Table 3, 3w).
| a Reaction conditions: amine (1.5 mmol), NCS (1.65 mmol, 1.1 equiv.), in 4 mL acetonitrile, stirring at room temperature for 3 h. To this reaction mixture were added benzyl alcohol (1.8 mmol, 1.2 equiv.), CuSO4·5H2O (20 mol%) and TBHP (7.5 mmol, 5 equiv.), stirring at 80 °C for 24 h. |
|---|
 |
In order to show the application of this new methodology, the Cu-catalyzed one-pot oxidative amidation was used as a key step to synthesis the biologically active N-acetylprocainamide 5, which was an antiarrhythmic agent, from commercial available 4-acetamidobenzyl alcohol 4 in 68% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of N-acetylprocainamide. | ||
On the basis of previous studies,7h,16 a possible mechanism was proposed as shown in Scheme 4. Firstly, alcohol is oxidized to aldehyde by TBHP. And Cu(II) reacts with TBHP to form a tert-butylperoxy radical, Cu(I) and H+ following the mechanism proposed in literature.17 Then the tert-butylperoxy radical captures hydrogen from aldehyde to generate an acyl radical, as reported by Wan.14 And the protonated N-chloramine is then converted into an amino radical by a redox reaction as well documented by Minisci.18 Finally, the acyl radical and the amino radical couple to form the desired amide. To confirm this hypothesis, the acyl radical, generated from benzyl alcohol under our reaction conditions, was trapped with 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), obtaining the TEMPO adduct, which was detected by HRMS (Scheme 5).
 | ||
| Scheme 4 Proposed mechanism for amide synthesis. | ||
 | ||
| Scheme 5 Trapping of the acyl radical. | ||
In conclusion, we have developed an efficient and green one-pot procedure for the synthesis of amides via copper-catalyzed direct oxidative amidation of aliphatic and aromatic alcohols with N-chloramines, prepared in situ from variously substituted amines. This method integrates alcohol oxidation and amide bond formation, which are usually accomplished separately, into a single operation. And the use of copper as a catalyst, TBHP as sole oxidant under mild reaction conditions makes this methodology novel and environmentally benign. More importantly, it has a wide substrate scope, and uses cheap, abundant, and easily available reagents.
Acknowledgements
The research has been supported by the National Natural Science Foundation of China (Grant No. 21522604, U1463201 and 21402240); the youth in Jiangsu Province Natural Science Fund (Grant No. BK20150031, BK20130913 and BY2014005-03); a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- Selected recent examples: (a) Y. J. Kang, H. A. Chung, J. J. Kim and Y. J. Yoon, Synthesis, 2002, 733 CrossRef CAS; (b) I. Azumaya, T. Okamoto, F. Imabeppu and H. Takayanagi, Tetrahedron Lett., 2003, 59, 2325 CrossRef CAS; (c) A. Teichert, K. Jantos, K. Harms and A. Studer, Org. Lett., 2004, 6, 3477 CrossRef CAS PubMed; (d) D. M. Shendage, R. Froehlich and G. Haufe, Org. Lett., 2004, 6, 3675 CrossRef CAS PubMed; (e) D. A. Black and B. A. Arndtsen, Org. Lett., 2006, 8, 1991 CrossRef CAS PubMed; (f) A. R. Katritzky, C. Cai and S. K. Singh, J. Org. Chem., 2006, 71, 3375 CrossRef CAS PubMed; (g) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) L. Yao and J. Aubé, J. Am. Chem. Soc., 2007, 129, 2766 CrossRef CAS PubMed; (b) R. F. Schmidt, Ber. Dtsch. Chem. Ges., 1924, 57, 704 CrossRef.
- (a) E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 89, 988 CrossRef; (b) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599 CrossRef CAS PubMed.
- I. Ugi, Angew. Chem., 1962, 74, 9 (Angew. Chem., Int. Ed. Engl., 1962, 1, 8) CrossRef CAS.
- Selected recent examples: (a) W.-J. Yoo and C.-J. J. Li, J. Am. Chem. Soc., 2006, 128, 13064 CrossRef CAS PubMed; (b) J. Gao and G.-U. Wang, J. Org. Chem., 2008, 73, 2955 CrossRef CAS PubMed; (c) C. Qian, X. Zhang, J. Li, F. Xu, Y. Zhang and Q. Shen, Organometallics, 2009, 28, 3856 CrossRef CAS; (d) J. M. Li, F. Xu, Y. Zhang and Q. Shen, J. Org. Chem., 2009, 74, 2575 CrossRef CAS PubMed; (e) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992 CrossRef PubMed; (f) S. C. Ghosh, J. S. Y. Ngiam, C. L. L. Chai, A. M. Seayad, D. T. Tuan and A. Chen, Adv. Synth. Catal., 2012, 354, 1407 CrossRef CAS; (g) C. G. Subhash, S. Y. N. Joyce, A. M. Seayad, T. T. Dang, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007 CrossRef PubMed; (h) C. Roberta, P. Andrea, G. Giampaolo and L. D. Luca, Org. Lett., 2012, 14, 5014 CrossRef PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed.
- (a) L. U. Nordstrom, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672 CrossRef CAS PubMed; (b) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667 CrossRef CAS PubMed; (c) Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374 CrossRef CAS; (d) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559 CrossRef CAS PubMed.
- (a) K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977 CrossRef CAS PubMed; (b) Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917 CrossRef CAS PubMed; (c) J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550 CrossRef PubMed.
- X.-F. Wu, M. Sharif, A. Pews-Davtyan, P. Langer, K. Ayub and M. Beller, Eur. J. Org. Chem., 2013, 2783 CrossRef CAS.
- X. Bantreil, C. Fleith, J. Martinez and F. Lamaty, ChemCatChem, 2012, 4, 1922 CrossRef CAS.
- X. Bantreil, N. Kanfar, N. Gehin, E. Golliard, P. Ohlmann, J. Martinez and F. Lamaty, Tetrahedron, 2014, 70, 5093 CrossRef CAS.
- Z. Liu, J. Zhang, S. Chen, E. Shi, Y. Xu and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231 CrossRef CAS PubMed.
- K. Xu, Y. Hu, S. Zhang, Z. Zha and Z. Wang, Chem.–Eur. J., 2012, 18, 9793 CrossRef CAS PubMed.
- S. Gaspa, A. Porcheddu and L. D. Luca, Org. Biomol. Chem., 2013, 11, 3803 CAS.
- F. Minisci, F. Fontana, S. Araneo, F. Recupero, S. Banfi and S. Quici, J. Am. Chem. Soc., 1995, 117, 226 CrossRef CAS.
- F. Minisci, Synthesis, 1973, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra. See DOI: 10.1039/c6ra20732d |
| This journal is © The Royal Society of Chemistry 2016 |