DOI:
10.1039/C6RA20725A
(Paper)
RSC Adv., 2016,
6, 102733-102743
CuO nanostructures of variable shapes as an efficient catalyst for [3 + 2] cycloaddition of azides with terminal alkyne†
Received
17th August 2016
, Accepted 21st October 2016
First published on 21st October 2016
Abstract
CuO nanostructures of variable shapes: CuO nanospheres (5–10 nm), CuO nanorods (W × L = 24–27 nm × 124–140 nm) and CuO nanowires (W × L = 8–10 nm × 230–270 nm) have been synthesised to study the effect of shape of the catalyst on the Cu(I)-catalysed “click” azide–alkyne cycloaddition. Cu(I) species were generated in situ by the reduction of CuO nanostructures in the presence of sodium ascorbate. CuO nanowires exhibited highest catalytic efficiency for the cycloaddition reaction between azide and terminal alkyne, featuring short reaction time, soft reaction conditions and complete regioselectivity. We have further extended the study by using azides with varying functional groups (–OCH3 and –NO2) and studied the effect of shape of the nanostructures on the rate of the reaction and yield of the triazole products. The activity trend observed was: CuO-NW > CuO-NR > CuO-NS, irrespective of the presence of electron withdrawing or donating groups on the azide.
Introduction
One-dimensional nanoparticles (NPs) [nanowires (NW), nanorods (NR), nanobelts (NB) etc.] with high surface area and small size have attracted remarkable attention in recent years due to their wide applications in catalysis.1 The catalytic activity and selectivity of the nanocatalysts is directly affected by their shape, as NPs with different shapes have different facets and different ratios of the number of atoms on the corners and edges to those on the facets.2 Furthermore, the nanoparticle catalysed reactions provide the advantages of high atom efficiency, simplified isolation of products, easy recovery and recyclability of the catalysts. Transition metal oxides NPs, predominantly cupric oxide (CuO), have attracted considerable attention in catalysis due to square planar coordination of Cu atom to the neighbouring oxygen atoms and a monoclinic crystal structure, unlike other 3d metal oxides.3 Moreover, CuO NPs are highly stable, non-toxic, environmental friendly and can be easily recycled. CuO NPs have been used as heterogeneous catalyst in many important chemical reactions such as nitro-aromatic reduction,4 oxidations of carbon monoxide,5 C-arylation,6 epoxidation of olefins7 and cross-coupling reactions.8 The present article is intended to use CuO nanostructures of variable shapes as catalyst in Cu(I)-catalysed Huisgen azide–alkyne cycloaddition.
Huisgen 1,3-dipolar cycloaddition9 of terminal alkynes and organic azides is a classical example of click chemistry, that delivers 1,2,3-triazole10 with high regioselectivity under Cu(I) catalysis as demonstrated by Sharpless11 and Meldal12 independently. Compounds containing 1,2,3-triazole moiety found extensive applications as energetic binders,13 anti-microbial coatings,14 chemical sensors,15 biodegradable polymers,16 pharmaceutical chemistry,17 bio-conjugation18 and peptide modifications.19
Traditionally, in copper catalysed azide–alkyne cycloaddition, Cu(I) salts were used directly or generated in situ by reduction of Cu(II) salts [e.g. Sharpless–Fokin catalyst (CuSO4/sodium ascorbate)].11b,20 The conventional catalysts though the most often used are associated with difficulties like high catalyst loading, difficulty in handling and catalyst recyclability and slow reaction rate etc. The use of Cu(I) or Cu(II) NPs as catalyst for the above reaction has been active lately with numerous review articles published in the past decade.21 Orgueira et al.22 and Rothenberg et al.23 have reported the pioneer work in this area by demonstrating the use of Cu(0) nanoclusters and NPs as catalyst in azide–alkyne cycloaddition. However, the catalyst cannot be recovered and reused due to the dissolution of NPs in the reaction mixture. In another report, Himo et al.24 have reported copper metal alone as a catalyst in cycloaddition reaction owing to long reaction time and high catalyst loading. Mixed Cu/CuO NPs have been used as catalyst for cycloaddition reaction by Molteni et al. in 2006.25 In 2013, Bai and Chen's group have successfully synthesised a triazole drug by using Cu2O NP as catalyst for azide–alkyne cycloaddition and further demonstrated that Cu2O NP are more efficient catalyst than CuSO4/sodium ascorbate.26 Recently, Astruc et al. have reported a highly efficient, recyclable amphiphilic dendrimer nanoreactor for part-per-million (ppm) Cu(I) catalysed azide-alkyne cycloaddition in water.27 Solid support catalysts have been explored by various research groups for enhanced efficiency and reusability of the catalyst.28 Various heterogeneous systems such as copper supported on soluble and insoluble polymers,29 zeolities,30 charcoal,31 silica-type material (SAB-15),32 graphene nanosheet33 and Amberlyst A-21 (ref. 34) etc. have been explored for catalysis in azide–alkyne cycloaddition and success have been achieved to develop highly efficient catalytic systems which work with considerably low amount of Cu(I) catalyst.32,35 The attachment of support imparts higher surface for the reaction and hence enhance the catalytic efficiency. An alternative approach is to have CuO nanostructures of suitable shapes having higher surface area to avoid the use of solid support for the catalyst. In 2010, Park and Song et al. have highlighted the effect of well-defined CuO NPs of variable shapes (urchin, hollow spheres, and hollow cubes) on the azide–alkyne cycloaddition and documented that CuO hollow spheres gave best activity owing to large surface area of the catalyst.36 There are enormous reports on the use of Cu(I) or Cu(II) NPs as catalyst in azide–alkyne cycloaddition reaction,21 and yet a comparative account of their catalytic activity as a function of shape is rarely investigated. In this regard, we plan to synthesise CuO nanostructures of variable shapes (spheres, rods and wires) with specifically exposed crystal planes, to evaluate the effect of shape of the catalyst on the Cu(I)-catalysed “click” azide–alkyne cycloaddition. Moreover, aryl azides bearing substituents with different electronic demands have been investigated. We further explored the catalyst reusability and tried to understand the mechanism of azide–alkyne cycloaddition catalysed by CuO nanostructures.
Results and discussion
Structural, optical and morphological characterisation of CuO nanostructures
Powder X-ray diffraction (XRD) patterns of CuO nanostructures are shown in Fig. 1 (panels a–c). XRD patterns indicated that all peaks indexed to monoclinic CuO (space group C2/c) crystal phase as per JPCDS Card no. 80-1268. Absence of peaks corresponding to Cu(OH)2 and Cu2O confirmed high purity of synthesised CuO NPs. Different intensities and broadness of XRD peaks observed for variable CuO nanostructures suggested differences in crystallite size of CuO-NS (∼5 nm), CuO-NR (∼10 nm) and CuO-NW (∼25 nm) as calculated by the Debye–Scherrer equation d = kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ using the average values of (0 0 2) (2θ = 35.5°) and (2 0 0) (2θ = 38.8°) planes, where k is the Debye–Scherrer constant (0.89), θ is diffraction angle and β is full width at half-maximum. Presence of strongest peaks at lower Miller-indexed (002) and (200) reflections indicated the preferential crystal planes of the CuO-NR and CuO-NW. The difference in intensity of related peaks for the variable CuO nanostructures may be due to texture effect as well as dimension/morphological alteration in certain direction. Similar results highlight the variation of morphology and growth direction leading to the change in intensity of the XRD peaks were reported by Singh et al.37
cosθ using the average values of (0 0 2) (2θ = 35.5°) and (2 0 0) (2θ = 38.8°) planes, where k is the Debye–Scherrer constant (0.89), θ is diffraction angle and β is full width at half-maximum. Presence of strongest peaks at lower Miller-indexed (002) and (200) reflections indicated the preferential crystal planes of the CuO-NR and CuO-NW. The difference in intensity of related peaks for the variable CuO nanostructures may be due to texture effect as well as dimension/morphological alteration in certain direction. Similar results highlight the variation of morphology and growth direction leading to the change in intensity of the XRD peaks were reported by Singh et al.37
 |
| | Fig. 1 Powder X-ray diffraction (XRD) patterns of (a) CuO-NS (b) CuO-NR and (c) CuO-NW. | |
The absorption spectra of CuO-NS (Fig. 2) showed a broad peak at 290 nm with absorption edge at 426 nm. The calculated band-gap for synthesised CuO-NS is Eg = 2 eV, which is much larger than the reported Eg value for bulk CuO (1.65 eV).38 The blue-shift in Eg value was attributed to the enhancement of quantum confinement effect resulting from decrease in size of the synthesised CuO-NS (5–10 nm). The broad absorption band observed in the absorption spectra was probably due to dominant surface-related defects for intra-gap states. Ovchinnikov et al.38 have reported the role of electronic defects due to the presence of dopants or valence defects (O vacancies and Cu+) in CuO NPs that results in the development of intra-band states. The red-shift observed in the absorption spectra of CuO-NR and CuO-NW (Fig. 2) was due to their large size as compare to CuO-NS.
 |
| | Fig. 2 UV-visible absorption spectra of CuO-NS, CuO-NR and CuO-NW. | |
Panels a–c of Fig. 3 shows typical transmission electron microscopy (TEM) images for CuO-NS, CuO-NR and CuO-NW. The CuO-NS are uniformly dispersed with average diameter of 5–10 nm. The TEM image of CuO-NR (Fig. 3b) shows sharp-topped rod like structures of width 24–27 nm and length 124–140 nm. The TEM image of CuO-NW indicates that the nanowires have width 8–10 nm with length 230–270 nm are randomly agglomerated as observed in Fig. 3c. To further confirm the purity of synthesised CuO NPs, energy-dispersive X-ray (EDX) analysis was carried out. The EDX spectra (Fig. 3d) of CuO NPs showed peaks corresponding to copper and oxygen only, indicating 100% purity of the synthesised CuO NPs. To confirm the valence state of copper present in the synthesised CuO nanostructures X-ray photoelectron spectroscopy (XPS) analysis were done. A high intensity peaks at 933.5 eV, 953.2 eV and satellite peaks could be assigned to the binding energies of Cu(II) (Fig. 4).39
 |
| | Fig. 3 TEM images of (a) CuO-NS (b) CuO-NR (c) CuO-NW (d) EDX spectra of CuO NPs. | |
 |
| | Fig. 4 The XPS spectra of synthesised CuO-NW and the inset shows the Cu 2p core level binding energy spectrum. | |
The observed Brunauer–Emmett–Teller (BET) surface area for CuO-NS, CuO-NR and CuO-NW were 41, 53 and 61 m2 g−1 respectively, which was in accordance with their difference in geometric dimension and surface morphology (Table 1).
Table 1 Summary of surface morphology and surface area of variable CuO nanostructures
| CuO nanostructures |
TEM size (nm) |
Surface area (m2 g−1) |
| CuO-NS |
5–10 |
41 |
| CuO-NR |
W × L = 24–27 × 124–140 |
53 |
| CuO-NW |
W × L = 8–10 × 230–270 |
61 |
Effect of shape of CuO nanostructures on azide–alkyne cycloaddition
CuO nanostructures of variable shapes (NS, NR and NW) have been used as catalyst in the cycloaddition reaction of azides and terminal alkyne (Scheme 1). Firstly, to standardised the reaction conditions, series of experiments were conducted on the model reaction between phenyl azide and phenylacetylene. Best yields were obtained in the presence of 5 mol% of catalyst (synthesised CuO nanostructures) loading and H2O/t-BuOH (3:1) as solvent at room temperature.
 |
| | Scheme 1 [3 + 2] cycloaddition of phenylacetylene (1) and azide (2a–c) catalysed by CuO nanostructures (NS, NR and NW). | |
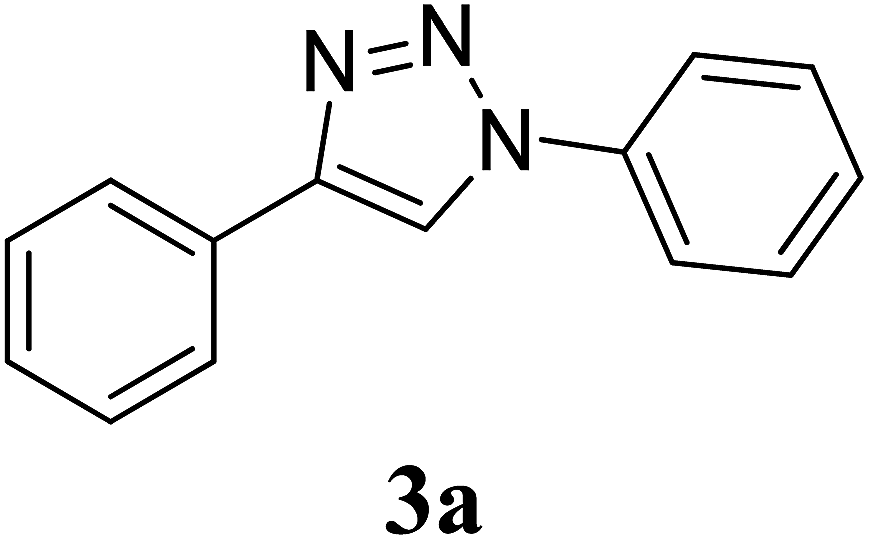
To investigate the effect of shape of CuO nanostructures on catalysis, phenylacetylene and phenyl azide were reacted in the presence of CuO-NS, CuO-NR and CuO-NW (Table 2, entry 1–3). The results indicated that the CuO-NW showed best catalytic activity yielding the desired triazole 3a as single regioisomer and in quantitative yield. Formation of compound 3a was confirmed by comparing its melting point (mp) and 1H NMR values with the reported data.40 The obtained results (Table 2, entry 1–3) for catalytic activity indicated that the anisotropy in the particle shape dominates over the isotropic particles and hence, CuO-NW and CuO-NR were found to be more active than CuO-NS. CuO-NW resulted in quantitative yield of 3a in 1 h due to much exposed surface active atoms. The relatively lower reactivity of CuO-NS as compared to CuO-NR and CuO-NW could be explained on the basis of effective surface area. The BET surface area of CuO nanostructures measured by nitrogen desorption experiments were 41, 53 and 61 m2 g−1 for CuO-NS, CuO-NR and CuO-NW respectively, and it was quite reasonable that the catalytic activities depend on the active surface area of the catalyst.
Table 2 [3 + 2] cycloaddition of phenylacetylene with various azides in the presence of CuO nanostructures of variable shapes under optimized reaction conditionsb
| Entry |
R |
Catalyst (5 mol%) |
Time (h) |
Product |
Yielda (%) |
| Yields were of isolated and purified products. Reaction conditions: 1.0 equiv. of alkyne, 1.1 equiv. of azide, 5 mol% catalyst, 0.1 equiv. sodium ascorbate in H2O/t-BuOH (3:1) at room temperature. |
| 1 |
H |
CuO-NS |
2 |
 |
85 |
| 2 |
H |
CuO-NR |
2 |
 |
89 |
| 3 |
H |
CuO-NW |
1 |
 |
100 |
| 4 |
OCH3 |
CuO-NS |
2 |
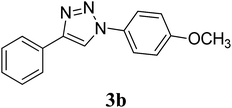 |
80 |
| 5 |
OCH3 |
CuO-NR |
1.5 |
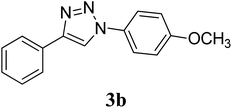 |
87 |
| 6 |
OCH3 |
CuO-NW |
1 |
 |
95 |
| 7 |
NO2 |
CuO-NS |
4 |
 |
40 |
| 8 |
NO2 |
CuO-NR |
4 |
 |
58 |
| 9 |
NO2 |
CuO-NW |
3 |
 |
67 |
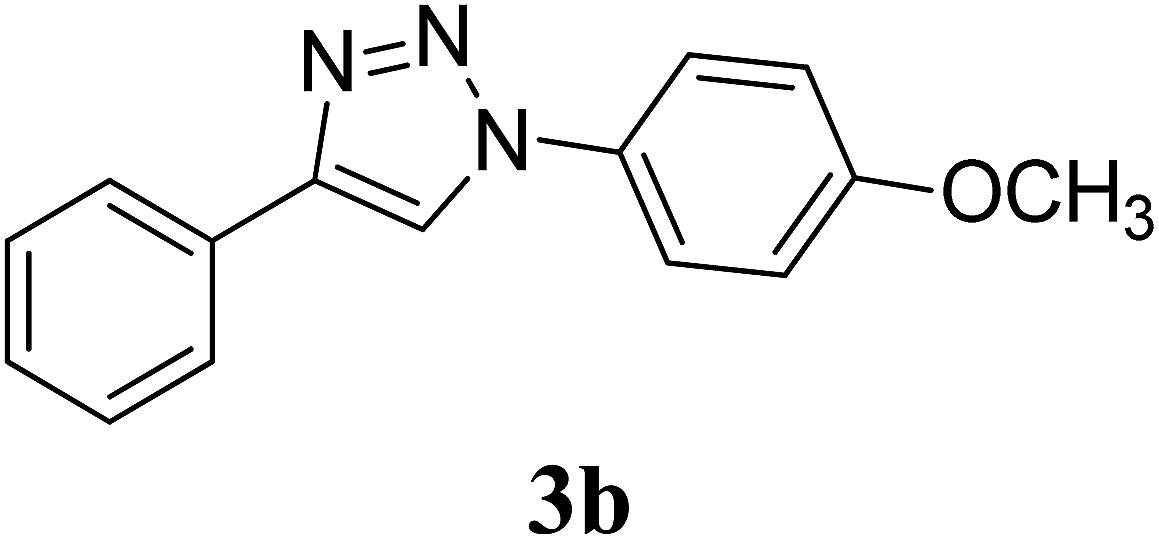
We further extended the study by using aryl azides bearing substituents with different electronic demands (–OCH3 and –NO2) and studied the effect of shape of the CuO nanostructures on the rate of reaction and yield of the triazole products (Table 2, entry 4–9). Observed results indicated that the desired triazole 3b was obtained in 95% yield in the presence of CuO-NW as catalyst, whereas the –NO2 substituted triazole 3c was obtained in comparatively less yield (67%), although CuO-NW provide best conversion as compared to CuO-NS and CuO-NR in all cases. The formation of compounds 3b40 and 3c41 was confirmed by comparing their mp and 1H NMR data with literature values. Overall, the activity trend observed was: CuO-NW > CuO-NR > CuO-NS, irrespective of the presence of electron withdrawing or donating groups on the azide.
Plausible catalytic pathway with synthesised CuO nanostructures
The plausible reaction pathway for the azide–alkyne cycloaddition has been shown in Scheme 2. In the first step, Cu(II) is reduce to Cu(I) in situ by sodium ascorbate. The Cu(I) species thus generated will react with an alkyne to give Cu–acetylide species A. The formation of the Cu–acetylide species A (greenish yellow colour) between the synthesised CuO-NW and alkyne in the presence of sodium ascorbate has been ascertain by infrared (IR) spectra (Fig. S1–S3†). The vibrational absorption of  at 1931 cm−1 (Lit.42 1930 cm−1, Lit.43 1940 cm−1, Lit.44 1936 cm−1) was observed in the heterogeneous composite of phenylacetylene, CuO-NW and sodium ascorbate. However, no such peak was observed in the absence of sodium ascorbate and the catalyst colour remains unchanged. These results indicate the formation of Cu–acetylide species A by the reaction of Cu(I) and alkyne. Similar IR studies were reported by Uozumi and coworkers with their catalyst to establish the formation of Cu–acetylide complex during Cu(I)-catalysed azide–alkyne cycloaddition reaction.44 In the XPS spectra of greenish yellow colour solid (Cu–acetylide species A) binding energy peaks appeared at a value different from those of CuO, suggesting that the new peaks could be assigned to Cu(I)–acetylide species (Fig. S4 and S5†).45 Later, the azide attack the Cu–acetylide A via 1,3-dipolar cycloaddition followed by protonation to yield the triazole and regeneration of the Cu(I) catalyst. Although, more recently various kinetic experiments21a,46 and computation studies47 have indicated the formation of bis(copper) complex as intermediate in the Cu(I) catalysed azide–alkyne cycloaddition. In a recent study, Bertrand and coworkers have isolated the mono- and bis-copper acetylide complexes and demonstrated that although both species are active in the catalytic cycle, the dinuclear complex is involved in the kinetically favoured pathway.48 The author's also highlighted the role of anionic ligands (LCuX; anionic ligand X = Cl, OAc, OPh. OTf etc.) in each individual step of Cu catalysed azide–alkyne cycloaddition reaction. Based on the postulated mechanism (Scheme 2), relatively low yield observed in case of –NO2 substituted azide (3c, 67%) could be justified. During the step c (Scheme 2) the azide coordinate with the complex A, the presence of electron withdrawing substituents on the azide will withdraw the electron density on the azide nitrogen and will decrease the reactivity.
at 1931 cm−1 (Lit.42 1930 cm−1, Lit.43 1940 cm−1, Lit.44 1936 cm−1) was observed in the heterogeneous composite of phenylacetylene, CuO-NW and sodium ascorbate. However, no such peak was observed in the absence of sodium ascorbate and the catalyst colour remains unchanged. These results indicate the formation of Cu–acetylide species A by the reaction of Cu(I) and alkyne. Similar IR studies were reported by Uozumi and coworkers with their catalyst to establish the formation of Cu–acetylide complex during Cu(I)-catalysed azide–alkyne cycloaddition reaction.44 In the XPS spectra of greenish yellow colour solid (Cu–acetylide species A) binding energy peaks appeared at a value different from those of CuO, suggesting that the new peaks could be assigned to Cu(I)–acetylide species (Fig. S4 and S5†).45 Later, the azide attack the Cu–acetylide A via 1,3-dipolar cycloaddition followed by protonation to yield the triazole and regeneration of the Cu(I) catalyst. Although, more recently various kinetic experiments21a,46 and computation studies47 have indicated the formation of bis(copper) complex as intermediate in the Cu(I) catalysed azide–alkyne cycloaddition. In a recent study, Bertrand and coworkers have isolated the mono- and bis-copper acetylide complexes and demonstrated that although both species are active in the catalytic cycle, the dinuclear complex is involved in the kinetically favoured pathway.48 The author's also highlighted the role of anionic ligands (LCuX; anionic ligand X = Cl, OAc, OPh. OTf etc.) in each individual step of Cu catalysed azide–alkyne cycloaddition reaction. Based on the postulated mechanism (Scheme 2), relatively low yield observed in case of –NO2 substituted azide (3c, 67%) could be justified. During the step c (Scheme 2) the azide coordinate with the complex A, the presence of electron withdrawing substituents on the azide will withdraw the electron density on the azide nitrogen and will decrease the reactivity.
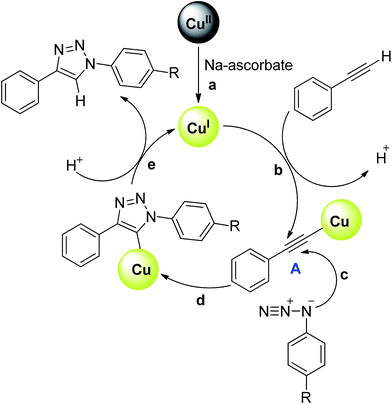 |
| | Scheme 2 Plausible catalytic pathway for alkyne–azide cycloaddition catalysed by CuO nanostructures. | |
Reusability study of the catalyst (CuO-NW)
The reusability of the catalyst (CuO-NW) was demonstrated by a set of experiments between phenylacetylene and p-methoxyphenyl azide (Table 3). After the first cycle, the catalyst was separated by centrifugation, washed with water and ethanol and dried in oven. Further, the recovered catalyst was used in a fresh reaction batch under the same reaction conditions. In a similar way the catalyst was recovered and used for at least 3 cycles without considerable loss in catalytic activity. The slight loss in catalytic activity may be due to loss of catalyst during washing, recovery process and leaching of the catalyst. To ascertain the amount of leached catalyst in the reaction mixture atomic absorption spectrophotometry (AAS) analysis was carried out. The AAS analysis showed that only 0.33 ppm of Cu leached after the reaction. To investigate whether the leached metal ions from the catalyst have contributed in catalytic activity, we have examined the reaction with or without catalyst. The click reaction was performed under optimized reaction condition but the catalyst was removed after 10 minutes by filtration. The reaction was again started and further conversion was monitored for another 3 hours. As observed in Fig. S6,† no significant gain in triazole yield was observed to rule out any considerable homogeneous contribution in catalytic activity. Thus, the reaction is essentially catalysed by solid catalyst and the leached metal ions did not contribute to catalytic activity.
Table 3 Reusability studies with CuO-NW as catalyst in cycloaddition reaction between phenylacetylene and p-methoxyphenyl azide under optimized reaction conditionsb
| Entry |
Run/cycle |
Product |
Time (h) |
Yielda (%) |
| Yields were of isolated and purified products. Reaction conditions: 1.0 equiv. of phenylacetylene, 1.1 equiv. of p-methoxy phenyl azide, 5 mol% catalyst, 0.1 equiv. sodium ascorbate in H2O/t-BuOH (3:1) at room temperature. |
| 1 |
1st run |
3b |
1 |
94 |
| 2 |
2nd run |
3b |
1 |
89 |
| 3 |
3rd run |
3b |
1.5 |
78 |
Conclusions
In conclusion, we have synthesised CuO nanostructures of variable shapes (CuO-NS, CuO-NR and CuO-NW) and demonstrated the effect of shape of CuO nanostructures on the catalytic efficiency in azide–alkyne cycloaddition. We observed that nanocatalysis is shape dependent and anisotropy in the particle shape showed dominance over the isotropic particles and hence CuO-NW and CuO-NR were found to be more active than the isotropic CuO-NS. The catalyst could be easily recovered and reused upto three cycles in azide–alkyne cycloaddition without considerable loss in catalytic efficiency.
Experimental section
Chemicals
Cupric nitrate [Cu(NO3)2·3H2O], cupric acetate [Cu(CH3COO)2], glacial acetic acid (CH3COOH), sodium hydroxide (NaOH), ethanol (CH3CH2OH) and aqueous ammonia, tertiary butanol, sodium ascorbate, phenylacetylene were purchased from Sigma Aldrich and used without further purification. Various aryl azides were prepared from the corresponding aryl amines by using the standardised literature procedure.49 De-ionized water (DI water) was obtained using an ultrafiltration system (Milli-Q, Milipore) with a measured conductivity of 35 mho cm−1 at 25 °C.
Synthesis of CuO nanostructures
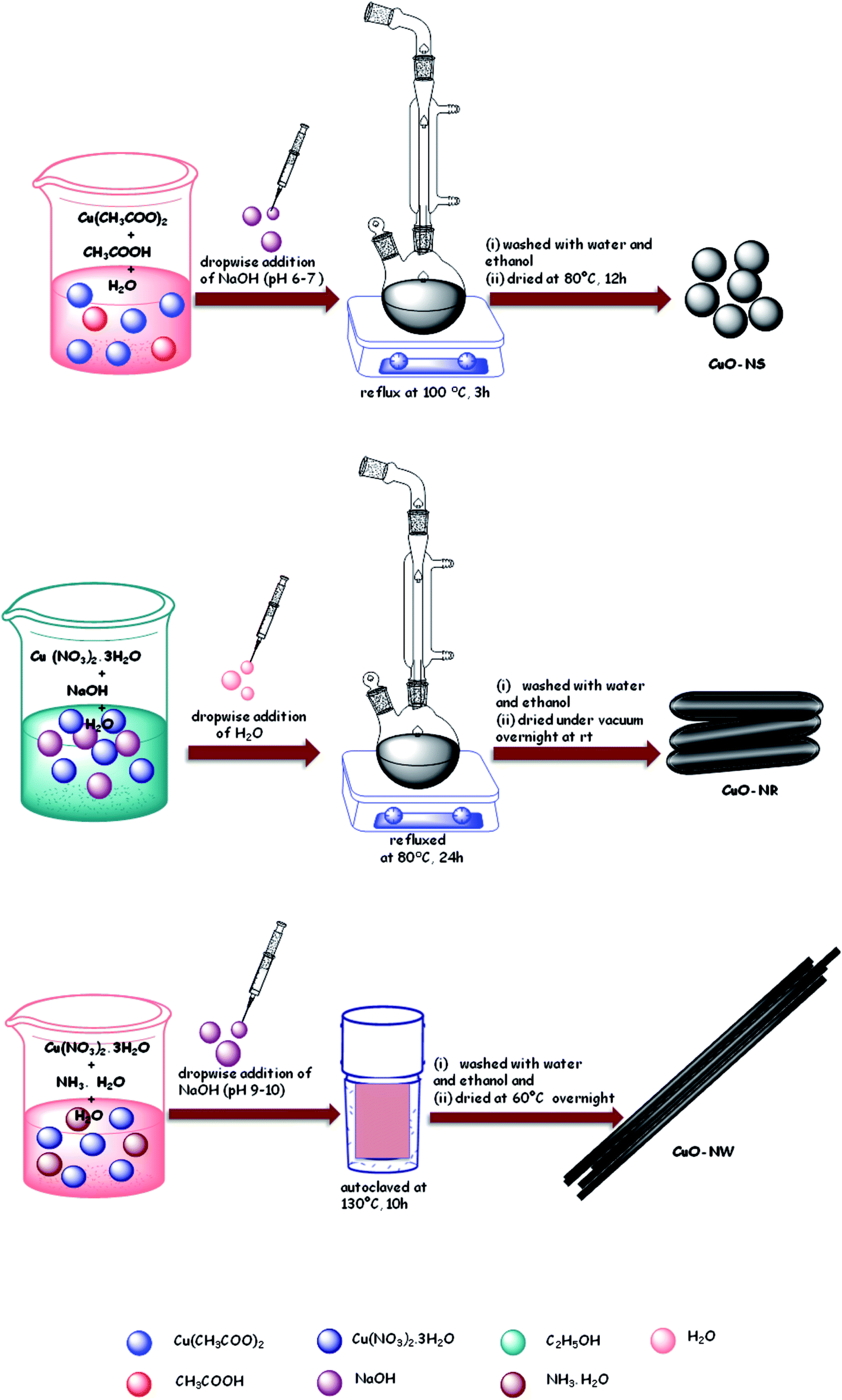
Schematic representation of synthesis of various CuO nanostructures (NS, NR and NW) has been shown in Scheme 3.
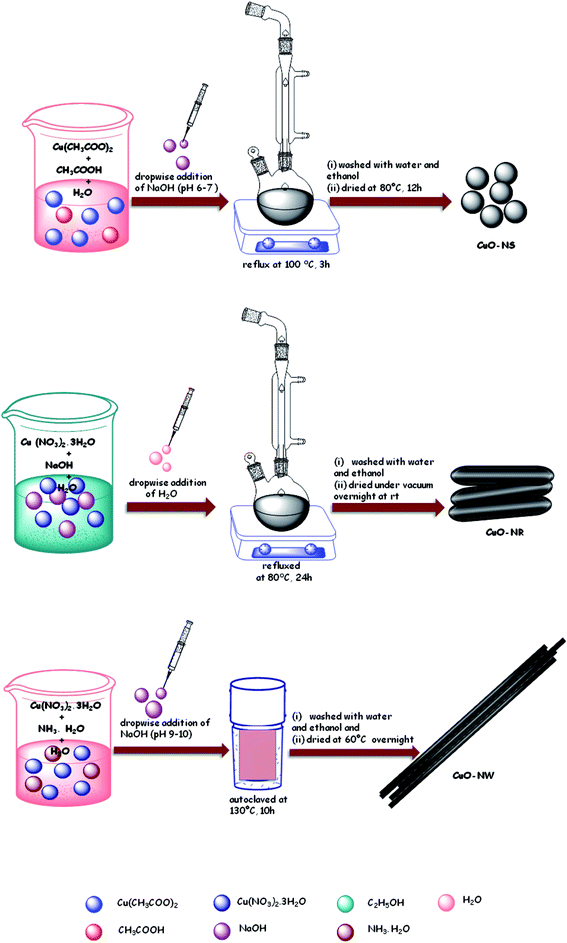 |
| | Scheme 3 Schematic representation for the synthesis of CuO-NS, CuO-NR and CuO-NW. | |
Synthesis of CuO-NS. In a round-bottomed flask, an aqueous solution of 0.02 M Cu(CH3COO)2 was mixed with 1 mL of glacial acetic acid and refluxed at 100 °C for 3 h with vigorous stirring. The reaction mixture was maintained at pH 6–7 with the addition of 0.1 M NaOH solution.50 On addition of NaOH black precipitates were formed and the reaction mixture was allowed to cool down. After cooling, the precipitates were centrifuged and washed with ethanol and water and dried in oven for 12 h.
Synthesis of CuO-NR. A 0.03 M solution of Cu(NO3)2·3H2O in 40 mL ethanol was added slowly into 40 mL ethanolic NaOH (0.5 M) solution in a round-bottomed flask and refluxed at 80 °C. After about 20 minutes, 60 mL of DI water was added to the above reaction mixture dropwise. The resultant product was refluxed together with mother liquor at 80 °C for 24 h.51 The black precipitates obtained were filtered and washed with DI water and ethanol several times; the solid sample was dried under vacuum overnight at room temperature.
Synthesis of CuO-NW. In a 100 mL DI water 1 g of Cu(NO3)2·3H2O was dissolved52 and then 30 mL aqueous ammonia solution was added with constant stirring. The pH of the above solution was maintained at 9–10 by dropwise addition of 1 M NaOH solution. The blue coloured Cu(OH)2 precipitate formed were filtered and washed several times with ethanol. The solid product was added to 30 mL DI water, sealed in a Teflon-lined stainless autoclave and kept at 130 °C for 10 h. Finally, black precipitates of CuO were washed with DI water and ethanol and dried at 60 °C overnight.
Characterisation of CuO-NS, CuO-NR and CuO-NW
The crystallographic information of the samples were investigated by powder X-ray diffraction (XRD) using a Panalytical's X'Pert Pro with Cu Kα (λ = 1.5406 Å) radiation. The absorbance spectra were recorded by UV-visible spectrophotometer (Perkin Elmer). The morphological characterisation has been performed by transmission electron microscopy (TEM) (Hitachi 7500 model). The X-ray photoelectron spectroscopy (XPS) measurements were carried out using a ESCA+ (Omicron nanotechnology, Oxford instrument Germany) equipped with monochromator Aluminium Source (Al Kα radiation; hν = 1486.7 eV). The instrument was operated at 15 kV and 20 mA with pass energy of 20 eV (for short scans) and 50 eV (for survey scans). The Brunauer–Emmett–Teller (BET) surface area was measured by (Smart Sorb 92–93) using 150 mg of sample preheated at 150 °C for 1 h. The metal ion concentration was estimated by atomic absorption spectrophotometry (AAS) (GBC 932 AA, Australia).
General experimental
All reactions were monitored by thin layer chromatography (TLC) using an appropriate solvent system for development. Yields reported are isolated yields. IR spectra were recorded with Bruker ALPHA FT-IR spectrometer. The coupling constants (J) are given in hertz (Hz) and chemical shifts are reported in parts per million (ppm). The abbreviations s, d, t and m refer to singlet, doublet, triplet and multiplet respectively. 1H NMR (500 MHz, CDCl3 or DMSO-d6) and 13C NMR (125 MHz, CDCl3 or DMSO-d6) spectra were recorded on a Bruker NMR spectrometer. HRMS data were recorded with electrospray ionization (ESI†) on Bruker MaXis Impact spectrometer. Melting points were recorded with a Perfit apparatus.
General procedure for [3 + 2] cycloaddition of azides and terminal alkynes catalysed by CuO nanostructures of variable shapes
A 25 mL round bottom flask was charged with CuO nanostructures (5 mol%), sodium ascorbate (0.1 equiv.), phenylacetylene (1.0 equiv.), and azide (1.1 equiv.) in H2O/t-BuOH (3:1). The resulting mixture was stirred at rt, until TLC indicated completion of reaction. The heterogenous catalyst was recovered by centrifugation. The mixture was diluted with water and ethyl acetate. The organic layer was separated, and the aqueous phase was extracted with ethyl acetate (2 × 10 mL). The combined organic layer was washed with aq NH4OH (0.2%) and brine, later dried over Na2SO4 and concentrated in vacuo to give the corresponding triazole product. The crude product was easily purified by flash chromatography on silica gel. We observed good conversions and yields for variety of azide–alkyne combinations (Table 2). The triazoles were characterized by 1H NMR, 13C NMR and HRMS.
Preparation of compound 3a. According to the general procedure, alkyne 1 (40 mg, 0.39 mmol), phenyl azide 2a (51.2 mg, 0.43 mmol), CuO nanostructures (NS/NR/NW) (1.5 mg, 0.02 mmol, 5 mol%) and sodium ascorbate (7.7 mg, 0.04 mmol) in H2O/t-BuOH (3:1) was stirred at rt till the completion of the reaction (2 h with CuO-NS, 2 h with CuO-NR and 1 h with CuO-NW respectively). The crude reaction mixture was purified by column chromatography (10% ethyl acetate/petroleum ether) to give the desired compound 3a as a white solid (73.3 mg. 85%) with CuO-NS, (76.8 mg, 89%) with CuO-NR and (86.3 mg, 100%) with CuO-NW respectively. Rf: 0.53 (20% ethyl acetate/petroleum ether). Mp: 171–173 °C (Lit.40 170–172 °C). 1H NMR (500 MHz, CDCl3): δ = 8.20 (s, 1H), 7.91 (d, J = 7.4 Hz, 2H), 7.79 (d, J = 7.9 Hz, 2H), 7.55 (t, J = 7.6 Hz, 2H), 7.47–7.45 (m, 3H), 7.37 (t, J = 7.4 Hz, 1H) ppm. 13C NMR (125 MHz, CDCl3): δ = 117.84, 120.72, 126.04, 128.62, 128.97, 129.11, 129.97, 130.39, 137.24 ppm. HRMS (ESI) m/z: calcd for C14H12N3 [M + H]+ 222.1026, found: 222.1026.
Preparation of compound 3b. According to the general procedure, alkyne 1 (40 mg, 0.39 mmol), p-methoxyphenyl azide 2b (63.9 mg, 0.43 mmol), CuO nanostructures (NS/NR/NW) (1.5 mg, 0.02 mmol, 5 mol%) and sodium ascorbate (7.7 mg, 0.04 mmol) in H2O/t-BuOH (3:1) was stirred at rt till the completion of the reaction (2 h with CuO-NS, 1.5 h with CuO-NR and 1 h with CuO-NW respectively). The crude mixture was purified by column chromatography (20% ethyl acetate/petroleum ether) to give the desired compound 3b as a white solid in (78.4 mg, 80%) with CuO-NS, (85.3 mg, 87%) with CuO-NR and (93.1 mg, 95%) with CuO-NW respectively. Rf: 0.32 (15% ethyl acetate/petroleum ether). Mp: 167–169 °C (Lit.40 167–168 °C). 1H NMR (500 MHz, CDCl3): δ = 8.11 (s, 1H), 7.89 (d, J = 7.15 Hz, 2H), 7.67 (d, J = 8.9 Hz, 2H), 7.44 (t, J = 7.45 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.02 (d, J = 8.9 Hz, 2H), 3.86 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ = 55.78, 114.93, 118.04, 122.31, 125.96, 128.48, 129.05, 130.47, 130.63, 148.33, 159.98 ppm. HRMS (ESI) m/z: calcd. for C15H14N3O [M + H]+ 252.1131, found: 252.1131.
Preparation of compound 3c. According to the general procedure, alkyne 1 (40 mg, 0.39 mmol), p-nitrophenyl azide 2c (70.5 mg, 0.43 mmol), CuO nanostructures (NS/NR/NW) (1.5 mg, 0.02 mmol, 5 mol%) and sodium ascorbate (7.7 mg, 0.04 mmol) in H2O/t-BuOH (3:1) was stirred at rt till the completion of the reaction (4 h with CuO-NS, 4 h with CuO-NR and 3 h with CuO-NW respectively). The crude mixture was purified by column chromatography (20% ethyl acetate/petroleum ether) to give the desired compound 3c as a pale yellow solid in (41.5 mg, 40%) with CuO-NS, (60.2 mg, 58%) with CuO-NR and (69.6 mg, 67%) with CuO-NW respectively. Rf: 0.46 (20% ethyl acetate/petroleum ether). Mp: 235–237 °C (Lit.41 236–238 °C). 1H NMR (500 MHz, DMSO-d6): δ = 9.48 (s, 1H), 8.50–8.48 (m, 2H), 8.27–8.25 (m, 2H), 7.96–7.95 (m, 2H), 7.53–7.51 (m, 2H), 7.42–7.39 (m, 1H) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 120.44, 121.00, 125.93, 126.14, 129.15, 129.59, 130.19 ppm. HRMS (ESI) m/z: calcd for C14H11N4O2 [M + H]+ 267.0877, found: 267.0877.
IR experiments for the detection of {L·Cu(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CC6H5)}
CC6H5)}
In a 10 mL round bottom flask, CuO-NW (12 mg, 0.12 mmol), phenylacetylene 1 (120 mg, 1.17 mmol), and sodium ascorbate (48 mg, 0.35 mmol) was added in H2O/t-BuOH (3:1) as solvent. The reaction vessel was stirred at room temperature for 30 min. The colour of the CuO-NW gradually changes from black to greenish yellow. The reaction mixture was filtered, and the resulting greenish yellow solid was washed with water and hexane and dried in vacuo to deliver the greenish yellow colour solid {L·Cu(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC6H5)}, which shows a vibrational absorption of Cu–CC at 1931 cm−1 (Lit.42 1930 cm−1, Lit.43 1940 cm−1, Lit.44 1936 cm−1). On the other hand, the reaction of CuO-NW, phenylacetylene 1 in the absence of sodium ascorbate didn't show any change in colour of the catalyst and also no peak was observed at 1930–1940 cm−1 in the IR spectrum.
CC6H5)}, which shows a vibrational absorption of Cu–CC at 1931 cm−1 (Lit.42 1930 cm−1, Lit.43 1940 cm−1, Lit.44 1936 cm−1). On the other hand, the reaction of CuO-NW, phenylacetylene 1 in the absence of sodium ascorbate didn't show any change in colour of the catalyst and also no peak was observed at 1930–1940 cm−1 in the IR spectrum.
Acknowledgements
D. G. and B. G. gratefully acknowledges Science and Engineering Research Board (SERB), Department of Science & Technology (DST), Government of India for the award of SERB Young Scientists Research Grant (Sanction No: SERB/F/4167/2015-16 and SB/FT/CS-013/2014). A. K. is thankful to SERB-DST, India for the award of fellowship. The authors acknowledge Department of Chemistry, Sri Guru Granth Sahib World University, Fatehgarh Sahib, Punjab, India for providing the research facilities.
References
-
(a) M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed;
(b) A. Gole and C. J. Murphy, Langmuir, 2008, 24, 266–272 CrossRef CAS PubMed;
(c) F. Teng, M. Chen, N. Li, X. Hua, K. Wang and T. Xu, ChemCatChem, 2014, 6, 842–847 CrossRef CAS;
(d) Q. Zhang, K. Zhang, D. Xu, G. Yang, H. Huang, F. Nie, C. Liu and S. Yang, Prog. Mater. Sci., 2014, 60, 208–337 CrossRef CAS;
(e) J. Liu, J. Jin, Z. Deng, S.-Z. Huang, Z.-Y. Hu, L. Wang, C. Wang, L.-H. Chen, Y. Li, G. V. Tendeloo and B.-L. Su, J. Colloid Interface Sci., 2012, 384, 1–9 CrossRef CAS PubMed;
(f) W. Zhang, Q. Zeng, X. Zhang, Y. Tian, Y. Yue, Y. Guo and Z. Wang, J. Org. Chem., 2011, 76, 4741–4745 CrossRef CAS PubMed;
(g) M. Suleiman, M. Mousa, A. Hussein, B. Hammouti, T. B. Hadda and I. Warad, J. Mater. Environ. Sci., 2013, 4, 792–797 CAS;
(h) R. Kaur and B. Pal, J. Mol. Catal. A: Chem., 2012, 355, 39–43 CrossRef CAS;
(i) F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2447–2448 RSC;
(j) Y. Jiang, X. He, W. Zhang, X. Li, N. Guo, Y. Zhao, G. Xu and W. Li, RSC Adv., 2015, 5, 73340–73345 RSC.
-
(a) R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2004, 126, 7194–7195 CrossRef CAS PubMed;
(b) R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- J. B. Forsyth and S. Hull, J. Phys.: Condens. Matter, 1991, 3, 5257–5261 CrossRef CAS.
- R. Kaur and B. Pal, Appl. Catal., A, 2015, 491, 28–36 CrossRef CAS.
- M. Y. Guo, F. Z. Liu, J. K. Tsui, A. A. Voskanyan, A. M. C. Ng, A. B. Djurišić, W. K. Chan, K. Y. Chan, C. Z. Liao, K. M. Shih and C. Surya, J. Mater. Chem. A, 2015, 3, 3627–3632 CAS.
- M. Kidwai, S. Bhardwaj and R. Poddar, Beilstein J. Org. Chem., 2010, 6, 35 Search PubMed.
- N. Scotti, N. Ravasio, F. Zaccheria, R. Psaro and C. Evangelisti, Chem. Commun., 2013, 49, 1957–1959 RSC.
-
(a) S. Jammi, S. Sakthivel, L. Rout, T. Mukherjee, S. Mandal, R. Mitra, P. Saha and T. Punniyamurthy, J. Org. Chem., 2009, 74, 1971–1976 CrossRef CAS PubMed;
(b) D. Alves, C. G. Santos, M. W. Paixão, L. C. Soares, D. de Souza, O. E. D. Rodrigues and A. L. Braga, Tetrahedron Lett., 2009, 50, 6635–6638 CrossRef CAS;
(c) H. Deol, S. Pramanik, M. Kumar, I. A. Khan and V. Bhalla, ACS Catal., 2016, 6, 3771–3783 CrossRef CAS.
- R. Huisgen, R. Knorr, L. Möbius and G. Szeimies, Chem. Ber., 1965, 98, 4014–4021 CrossRef CAS.
- B. Schulzeab and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS;
(b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- R. Reed Jr, US Pat., 6103029 A, 2000.
-
(a) S. Kantheti, R. Narayan and K. V. S. N. Raju, RSC Adv., 2015, 5, 3687–3708 RSC;
(b) N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC.
-
(a) B. G. De Geest, W. Van Camp, F. E. Du Prez, S. C. De Smedt, J. Demeester and W. E. Hennink, Macromol. Rapid Commun., 2008, 29, 1111–1118 CrossRef CAS;
(b) L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
-
(a) J.-F. Lutz, H. G. Börner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS;
(b) E. P. Lundberg, C. Plesa, L. M. Wilhelmsson, P. Lincoln, T. Brown and B. Nordén, ACS Nano, 2011, 5, 7565–7575 CrossRef CAS PubMed;
(c) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
-
(a) W. Tanga and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC;
(b) S. Kotha, D. Goyal, S. Banerjee and A. Datta, Analyst, 2012, 137, 2871–2875 RSC;
(c) S. Kotha, D. Goyal and A. S. Chavan, J. Org. Chem., 2013, 78, 12288–12313 CrossRef CAS PubMed;
(d) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed;
(e) S. Kotha, D. Goyal, A. Bitra, N. Thota, G. Kruger and R. Anand, RSC Adv., 2013, 3, 24447–24454 RSC.
-
(a) L. V. Lee, M. L. Mitchell, S. J. Huang, V. V. Fokin, K. B. Sharpless and C. H. Wong, J. Am. Chem. Soc., 2003, 125, 9588–9589 CrossRef CAS PubMed;
(b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
-
(a) C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS;
(b) T. Jin, M. Yan and Y. Yamamoto, ChemCatChem, 2012, 4, 1217–1229 CrossRef CAS;
(c) F. Alonso, Y. Moglie and G. Radivoy, Acc. Chem. Res., 2015, 48, 2516–2528 CrossRef CAS PubMed;
(d) H. Woo, H. Kang, A. Kim, S. Jang, J. C. Park, S. Park, B.-S. Kim, H. Song and K. H. Park, Molecules, 2012, 17, 13235–13252 CrossRef CAS PubMed.
- H. A. Orgueira, D. Fokas, Y. Isome, P. C.-M. Chan and C. M. Baldino, Tetrahedron Lett., 2005, 46, 2911–2914 CrossRef CAS.
- L. D. Pachón, J. H. van Maarseveen and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 811–815 CrossRef.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- G. Molteni, C. L. Bianchi, G. Marinoni, N. Santo and A. Ponti, New J. Chem., 2006, 30, 1137–1139 RSC.
- J.-H. Wang, C.-W. Pan, Y.-T. Li, F.-F. Meng, H.-G. Zhou, C. Yang, Q. Zhang, C.-G. Bai and Y. Chen, Tetrahedron Lett., 2013, 54, 3406–3409 CrossRef CAS.
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed.
-
(a) N. Joshi and S. Banerjee, Tetrahedron Lett., 2015, 56, 4163–4169 CrossRef CAS;
(b) R. N. Dhital, C. Kamonsatikul, E. Somsook, K. Bobuatong, M. Ehara, S. Karanjit and H. Sakurai, J. Am. Chem. Soc., 2012, 134, 20250–20253 CrossRef CAS PubMed;
(c) L. Gu, D. Ma, S. Yao, C. Wang, W. Shen and X. Bao, Chem. Commun., 2010, 46, 1733–1735 RSC;
(d) Y. Zhu, Z. Hua, X. Zhou, Y. Song, Y. Gong, J. Zhou, J. Zhao and J. Shi, RSC Adv., 2013, 3, 4193–4198 RSC;
(e) A. Schätz, T. R. Long, R. N. Grass, W. J. Stark, P. R. Hanson and O. Reiser, Adv. Funct. Mater., 2010, 20, 4323–4328 CrossRef PubMed;
(f) B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed;
(g) B. J. Borah, D. Dutta, P. P. Saikia, N. C. Barua and D. K. Dutta, Green Chem., 2011, 13, 3453–3460 RSC;
(h) B. Dervaux and F. E. Du Prez, Chem. Sci., 2012, 3, 959–966 RSC;
(i) P. Veerakumar, M. Velayudham, K.-L. Lu and S. Rajagopal, Catal. Sci. Technol., 2011, 1, 1512–1525 RSC.
-
(a) A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334–3340 CrossRef CAS;
(b) U. Sirion, Y. J. Bae, B. S. Lee and D. Y. Chi, Synlett, 2008, 2326–2330 CAS;
(c) R. B. N. Baig and R. S. Varma, Green Chem., 2013, 15, 1839–1843 RSC;
(d) F. Chahdoura, C. Pradel and M. Gómez, ChemCatChem, 2014, 6, 2929–2936 CrossRef CASM. Gholinejad and N. Jeddi, ACS Sustainable Chem. Eng., 2014, 2, 2658–2665 CrossRef CAS;
(e) M. Biswas, A. Saha, M. Dule and T. K. Mandal, J. Phys. Chem. C, 2014, 118, 22156–22165 CrossRef CAS.
- S. Chassaing, A. S. S. Sido, A. Alix, M. Kumarraja, P. Pale and J. Sommer, Chem.–Eur. J., 2008, 14, 6713–6721 CrossRef CAS PubMed.
-
(a) B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed;
(b) B. R. Buckley, R. Butterworth, S. E. Dann, H. Heaney and E. C. Stubbs, ACS Catal., 2015, 5, 793–796 CrossRef CAS.
- S. Roya, T. Chatterjeec, M. Pramanikd, A. S. Roy, A. Bhaumik and S. M. Islam, J. Mol. Catal. A: Chem., 2014, 386, 78–85 CrossRef.
- A. S. Nia, S. Rana, D. Dohler, F. Jirsa, A. Meister, L. Guadagno, E. Koslowski, M. Bron and W. H. Binder, Chem.–Eur. J., 2015, 21, 10763–10770 CrossRef PubMed.
-
(a) R. Slimi, S. Kalhor-Monfared, B. Plancq and C. Girard, Tetrahedron Lett., 2015, 56, 4339–4344 CrossRef CAS;
(b) C. D. Smith, I. R. Baxendale, S. Lanners, J. J. Hayward, S. C. Smith and S. V. Ley, Org. Biomol. Chem., 2007, 5, 1559–1561 RSC.
-
(a) L. Liang, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2011, 353, 3434–3450 CrossRef CAS;
(b) C. Wang, R. Ciganda, L. Salmon, D. Gregurec, J. Irigoyen, S. Moya, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2016, 55, 3091–3095 CrossRef CAS PubMed;
(c) C. Wang, D. Wang, S. Yu, T. Cornilleau, J. Ruiz, L. Salmon and D. Astruc, ACS Catal., 2016, 6, 5424–5431 CrossRef;
(d) A. Pourjavadia, N. Safaiea, S. H. Hosseinia and C. Bennett, Appl. Organomet. Chem., 2015, 29, 601–607 CrossRef.
- J. Y. Kim, J. C. Park, H. Kang, H. Song and K. H. Park, Chem. Commun., 2010, 46, 439–441 RSC.
- D. P. Singh, A. K. Ojha and O. N. Srivastava, J. Phys. Chem. C, 2009, 113, 3409–3418 CAS.
- S. G. Ovchinnikov, B. A. Gizhevskii, Y. P. Sukhorukov, A. E. Ermakov, M. A. Uimin, E. A. Kozlov, Y. Kotov and A. A. V. Bagazeev, Phys. Solid State, 2007, 49, 1116–1120 CrossRef CAS.
-
(a) D. A. Zatsepin, V. R. Galakhov, M. A. Korotin, V. V. Fedorenko, E. Z. Kurmaev, S. Bartkowski, M. Neumann and R. Berger, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 4377–4381 CrossRef CAS;
(b) J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics, Minnesota, 1995 Search PubMed.
- B. Kaboudin, R. Mostafalu and T. Yokomatsu, Green Chem., 2013, 15, 2266–2274 RSC.
- C. V. Ramana, S. Chatterjee, K. A. Durugkar and R. G. Gonnade, CrystEngComm, 2009, 11, 143–150 RSC.
- C. Shao, G. Cheng, D. Su, J. Xu, X. Wang and Y. Hu, Adv. Synth. Catal., 2010, 352, 1587–1592 CrossRef CAS.
- W. M. Khairul, M. A. Fox, N. N. Zaitseva, M. Gaudio, D. S. Yufit, B. W. Skelton, A. H. White, J. A. K. Howard, M. I. Bruce and P. J. Low, Dalton Trans., 2009, 610–620 RSC.
- Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 9285–9290 CrossRef CAS PubMed.
- T. Jin, M. Yan, Menggenbateer, T. Minato, M. Bao and Y. Yamamoto, Adv. Synth. Catal., 2011, 353, 3095–3100 CrossRef CAS.
-
(a) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC;
(b) A. N. Semakin, D. P. Agababyan, S. Kim, S. Lee, A. Y. Sukhorukov, K. G. Fedina, J. Oh and S. L. Ioffe, Tetrahedron Lett., 2015, 56, 6335–6339 CrossRef CAS;
(c) V. O. Rodionov, S. I. Presolski, S. Gardinier, Y.-H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696–12704 CrossRef CAS PubMed;
(d) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210–2215 CrossRef CAS PubMed.
- M. Ahlquist and V. V. Fokin, Organometallics, 2007, 26, 4389–4391 CrossRef CAS.
- L. Jin, E. A. Romero, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 15696–15698 CrossRef CAS PubMed.
- C. Gill, G. Jadhav, M. Shaikh, R. Kale, A. Ghawalkar, D. Nagargoje and M. Shiradkar, Bioorg. Med. Chem. Lett., 2008, 18, 6244–6247 CrossRef CAS PubMed.
- J. Zhu, D. Li, H. Chen, X. Yang, L. Lu and X. Wang, Mater. Lett., 2004, 58, 3324–3327 CrossRef CAS.
- Y. Chang and H. C. Zeng, Cryst. Growth Des., 2004, 4, 397–402 CAS.
- G. H. Du and G. V. Tendeloo, Chem. Phys. Lett., 2004, 393, 64–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H/13C NMR and HRMS spectra of synthesised compounds and Fig. S1–S6. See DOI: 10.1039/c6ra20725a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Sukhmani Mann,
Bhupesh Goyal
,
Sukhmani Mann,
Bhupesh Goyal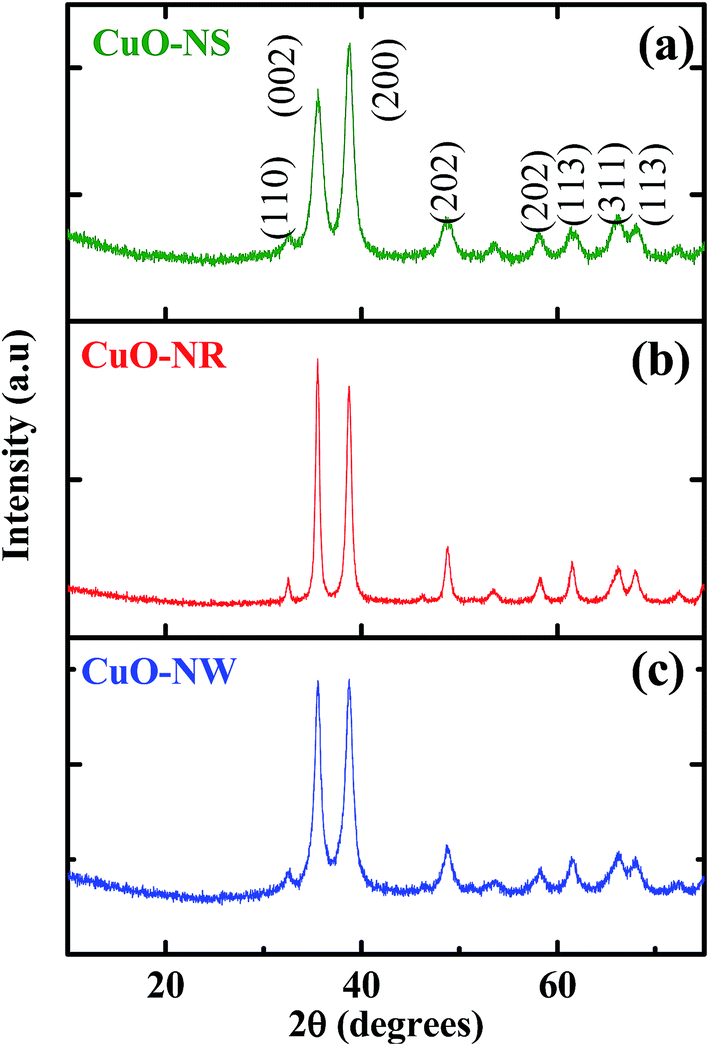











 at 1931 cm−1 (Lit.42 1930 cm−1, Lit.43 1940 cm−1, Lit.44 1936 cm−1) was observed in the heterogeneous composite of phenylacetylene, CuO-NW and sodium ascorbate. However, no such peak was observed in the absence of sodium ascorbate and the catalyst colour remains unchanged. These results indicate the formation of Cu–acetylide species A by the reaction of Cu(I) and alkyne. Similar IR studies were reported by Uozumi and coworkers with their catalyst to establish the formation of Cu–acetylide complex during Cu(I)-catalysed azide–alkyne cycloaddition reaction.44 In the XPS spectra of greenish yellow colour solid (Cu–acetylide species A) binding energy peaks appeared at a value different from those of CuO, suggesting that the new peaks could be assigned to Cu(I)–acetylide species (Fig. S4 and S5†).45 Later, the azide attack the Cu–acetylide A via 1,3-dipolar cycloaddition followed by protonation to yield the triazole and regeneration of the Cu(I) catalyst. Although, more recently various kinetic experiments21a,46 and computation studies47 have indicated the formation of bis(copper) complex as intermediate in the Cu(I) catalysed azide–alkyne cycloaddition. In a recent study, Bertrand and coworkers have isolated the mono- and bis-copper acetylide complexes and demonstrated that although both species are active in the catalytic cycle, the dinuclear complex is involved in the kinetically favoured pathway.48 The author's also highlighted the role of anionic ligands (LCuX; anionic ligand X = Cl, OAc, OPh. OTf etc.) in each individual step of Cu catalysed azide–alkyne cycloaddition reaction. Based on the postulated mechanism (Scheme 2), relatively low yield observed in case of –NO2 substituted azide (3c, 67%) could be justified. During the step c (Scheme 2) the azide coordinate with the complex A, the presence of electron withdrawing substituents on the azide will withdraw the electron density on the azide nitrogen and will decrease the reactivity.
at 1931 cm−1 (Lit.42 1930 cm−1, Lit.43 1940 cm−1, Lit.44 1936 cm−1) was observed in the heterogeneous composite of phenylacetylene, CuO-NW and sodium ascorbate. However, no such peak was observed in the absence of sodium ascorbate and the catalyst colour remains unchanged. These results indicate the formation of Cu–acetylide species A by the reaction of Cu(I) and alkyne. Similar IR studies were reported by Uozumi and coworkers with their catalyst to establish the formation of Cu–acetylide complex during Cu(I)-catalysed azide–alkyne cycloaddition reaction.44 In the XPS spectra of greenish yellow colour solid (Cu–acetylide species A) binding energy peaks appeared at a value different from those of CuO, suggesting that the new peaks could be assigned to Cu(I)–acetylide species (Fig. S4 and S5†).45 Later, the azide attack the Cu–acetylide A via 1,3-dipolar cycloaddition followed by protonation to yield the triazole and regeneration of the Cu(I) catalyst. Although, more recently various kinetic experiments21a,46 and computation studies47 have indicated the formation of bis(copper) complex as intermediate in the Cu(I) catalysed azide–alkyne cycloaddition. In a recent study, Bertrand and coworkers have isolated the mono- and bis-copper acetylide complexes and demonstrated that although both species are active in the catalytic cycle, the dinuclear complex is involved in the kinetically favoured pathway.48 The author's also highlighted the role of anionic ligands (LCuX; anionic ligand X = Cl, OAc, OPh. OTf etc.) in each individual step of Cu catalysed azide–alkyne cycloaddition reaction. Based on the postulated mechanism (Scheme 2), relatively low yield observed in case of –NO2 substituted azide (3c, 67%) could be justified. During the step c (Scheme 2) the azide coordinate with the complex A, the presence of electron withdrawing substituents on the azide will withdraw the electron density on the azide nitrogen and will decrease the reactivity.