
Carbon dioxide activation and transformation to HCOOH on metal clusters (M = Ni, Pd, Pt, Cu, Ag & Au) anchored on a polyaniline conducting polymer surface – an evaluation study by hybrid density functional theory†
Abstract
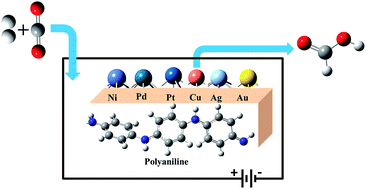
Developing new efficient catalysts for the electrochemical reduction of carbon dioxide to formic acid is important in the process of mitigating environmental CO2. In the present work, we have designed metal (M) clusters anchored on a polyaniline (PANI) conducting polymer electrode (M@PANI), where, M = Ni, Pd, Pt, Cu, Ag & Au, and evaluated their potential catalytic activity towards CO2 reduction by means of computational hydrogen electrode using hybrid density functional theory methods. The predicted binding energy and electronic properties of M@PANI suggest a thermodynamically feasible reaction which retains its conducting property with enhancement. The modified electrodes favour the formation of HCOOH involving H*COO species via the formate pathway. The computed limiting potentials suggest that Cu@PANI is a suitable electrode material for the CO2 reduction reaction leading to HCOOH.
 Please wait while we load your content...
Please wait while we load your content...

