DOI:
10.1039/C6RA20702B
(Paper)
RSC Adv., 2016,
6, 87246-87257
Mechanistic study of visible light driven photocatalytic degradation of EDC 17α-ethinyl estradiol and azo dye Acid Black-52: phytotoxicity assessment of intermediates†
Received
17th August 2016
, Accepted 31st August 2016
First published on 1st September 2016
Abstract
The present study evaluated the phytotoxicity of degraded intermediates of an endocrine-disrupting compound (EDC) 17α-ethinyl estradiol (EE2) and toxic azo dye Acid Black-52 (AB-52) in photocatalytic degradation. A novel bimetallic (silver and zirconium) doped TiO2 nanoparticle was synthesized for utilization in degrading these pollutants under visible light. The degradation pathway of the pollutants during photocatalytic activity was investigated by LC-MS analysis. Further, to understand the toxicity of intermediate compounds compared with the pollutants, phytotoxicity was assessed on two different seeds, Vigna radiata and Phaseolus vulgaris. Seeds treated with 100 ppm concentration of AB-52 showed low germination percentage in V. radiata (30%), and P. vulgaris (40%). Similarly, seeds treated with EE2 also showed less germination in V. radiata (40%), and P. vulgaris (50%) compared to intermediate compounds (100% germination) and revealed the less toxic nature of the degraded metabolites compared to EE2 and AB-52. Further, active radical scavenging experiments were carried out to understand the main species involved in the photocatalytic degradation process. A photoluminescence study and reactive oxygen species generation results suggested that the efficient charge carrier separation took place during the irradiation. Thus, the present work proves the ability of effective multifunctional nanomaterials to not only to degrade hazardous pollutants but also to detoxify them.
Introduction
The major cause of progressive and increased levels of pharmaceutical contaminants in water is their large production and utilization all over the world (ca. 4000 types in the order of 100–200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year).1,2 Most of the drugs that are prescribed and consumed by humans and animals are later excreted and finally end up in wastewater treatment plants (WWTPs). Therefore, it is of worldwide interest to minimize these pollutants since they can be very toxic to ecosystems even at very low concentrations (ppb). Many of these pharmaceutical ingredients (APIs) are intentionally made to be stable so as to remain biologically active with the aim of being effective in health care. Therefore, the residue of these drugs is very difficult to treat by conventional processes applied in WWTPs, because only a fraction of them are being destroyed.3 As a result, higher levels of these emerging pollutants have been detected in aquatic environments and even in drinking water during the last few years. US EPA pronouncements define these emerging contaminants as chemical compounds which are not modulated and whose effect on the environment and human health is poorly understood as yet.4
000 tons per year).1,2 Most of the drugs that are prescribed and consumed by humans and animals are later excreted and finally end up in wastewater treatment plants (WWTPs). Therefore, it is of worldwide interest to minimize these pollutants since they can be very toxic to ecosystems even at very low concentrations (ppb). Many of these pharmaceutical ingredients (APIs) are intentionally made to be stable so as to remain biologically active with the aim of being effective in health care. Therefore, the residue of these drugs is very difficult to treat by conventional processes applied in WWTPs, because only a fraction of them are being destroyed.3 As a result, higher levels of these emerging pollutants have been detected in aquatic environments and even in drinking water during the last few years. US EPA pronouncements define these emerging contaminants as chemical compounds which are not modulated and whose effect on the environment and human health is poorly understood as yet.4
The widespread presence of PPCPs in the environment has been extensively reported in order to increase global recognition and action.5,6 Among PPCPs, 17α-ethinylestradiol (EE2), the synthetic estrogen and endocrine-disrupting chemical (EDC) is ranked in the top 100 of priority PPCPs.7 EE2 is a primary constituent in contraceptive pills and postmenopausal hormonal appurtenances and is more fractious than normal estrogen in terms of its removal during water treatment.8 EE2 is often found in wastewater treatment plants (WWTPs) and is discharged into receiving waters due to incomplete removal during the treatment process.9 Although it is present in trace amounts (parts-per-billion level), EE2 can induce the feminization of male fish and alter the reproductive potential of the fish population.10 Due to the adverse effects of EE2 on ecosystems, human health and drinking water safety, and the ineffective EE2 removal from WWTPs, it is essential to explore effective techniques for EE2 removal within WWTPs.
Synthetic organic (azo) dyes have been utilized extensively in textile, cosmetic, paper, drug and food processing industries. These synthetic colorants are not easily degradable by biological treatment methods, due to their complex structure and high refractoriness. The majority of these dyes are highly carcinogenic and harmful and have the capacity to reduce light penetration in aqueous systems; thus they have a negative effect on photosynthesis, apart from their effect on human health.11 The dye-containing effluents are determined by their fluctuating pH, suspended particles, high oxygen demand, non-biodegradability and resistance to oxidation. Moreover, formal water decontamination methods are often chemically, energetically and operationally suitable only for large systems12,13 thereby demanding the development of new materials for effective dye removal.
The aim of the present study is to evaluate the potential of metal-doped TiO2 nanoparticles to degrade hazardous toxicants 17α-ethinyl estradiol and Acid Black-52. The transformation pathway and intermediate compounds have been identified through LCMS analysis during the degradation process. Phytotoxicity assessment upon two common plant seeds Vigna radiata and Phaseolus vulgaris indicated the less toxic nature of the degraded metabolites compared to toxicants 17α-ethinyl estradiol and Acid Black-52.
Materials and methods
Materials
Titanium(IV) isopropoxide, terepthalic acid (TA), nitro blue tetrazolium chloride (NBT) and 17α-ethinyl estradiol (EE2) from Sigma-Aldrich, Acid Black-52 dye from a local textile industry, hydrazine hydrate, zirconyl nitrate [ZrO(NO3)2], isopropanol, potassium iodide (KI), EDTA, potassium dichromate (K2Cr2O7) ascorbic acid, ethanol and Tween 20 from Alibaba Chemicals, China were used in the present study.
Synthesis of metal-doped TiO2 nanoparticles
TiO2 nanoparticles were prepared using the following method; a mixture of 5 mL of titanium(IV) isopropoxide in 50 mL isopropanol was added drop wise to 200 mL of distilled water maintained at pH 1.5 while the solution was continuously stirred. This TiO2 sol was dried at 100 °C for 24 h, and then calcined at 450 °C for 4 h to obtain the nanoparticles. Ag/TiO2 and Zr/Ag–TiO2 nanoparticles were prepared by the above method with a few modifications. For preparation of Ag–TiO2 nanoparticles, a required amount of aqueous solution of AgNO3 (0.2–0.8 mol%), while for Zr/Ag–TiO2 nanoparticles required amounts of aqueous solutions of both AgNO3 and ZrO(NO3)2 (0.2–0.8 mol%) were added drop wise to the TiO2 sol with continuous stirring for 45 min. A small aliquot of distilled water, 0.05 M hydrazine hydrate and 5 mL of Tween 20 were added to all the above solutions with continuous stirring for an additional 30 min. The resultant sol was sonicated at 80 MHz for 90 min and then dried at 100 °C in a hot air oven for 24 h to obtain the dry gel. The gel was then calcinated at 450 °C to obtain the required nanoparticle powders.
Characterization of nanoparticles
A powder XRD crystallogram was recorded using an X-ray BRUKER D8 Advance X-ray diffractometer with a Cu Kα source (λ = 1.5406 Å). The crystalline phase of the nanoparticles was identified by comparing the major peak positions with standard JCPDS files. A JEOL JEM 2100 High Resolution Transmission Electron Microscope (HRTEM) was used for imaging, SAED pattern and energy dispersive X-ray pattern with an accelerating voltage of 200 kV at different magnifications. Diffuse reflectance spectra were recorded using a JASCO V-670 UV-Vis spectrophotometer. The photoluminescence (PL) spectra were obtained using a HITACHI F-7000 fluorescence spectrophotometer. XPS data was acquired using a Kratos Axis Ultra 165 Spectrometer with a monochromated Al Kα X-ray source (hα = 1486.6 eV).
Photocatalytic degradation of EE2 and AB-52
All photodegradation experiments were carried out using a multitube photoreactor (Fig. 1). The photoreactor consists of a large borosil tube in the middle of the reactor facilitated with water inlet and outlet in order to avoid temperature fluctuation during the degradation process. Surrounding the central tube, six tubes (50 mL) were equipped with an air pump to maintain a constant mixing of the catalyst with the effluent solution. A 300 W Xe lamp (λ ≥ 400 nm) was utilized for the photodegradation experiments. This multitube reactor has been utilized for simultaneous degradation of both EE2 and AB-52. Initially the required quantity of EE2 and catalyst was taken in a glass beaker, and then stirred in the dark for 1 h to ensure absorption–desorption equilibrium.14,15 The leftover concentration of EE2 was determined using a High Performance Liquid Chromatography (HPLC) technique. An Agilent 1260 series with an Eclipse XDBC18 (5 mm) reverse phase column (4.6 × 150 mm) was used for the separation. A fluorescence detector with an excitation wavelength of 280 nm and an emission wavelength of 310 nm was used in this study.16 The mobile phase consisted of a mixture of water and methanol (30:70 v/v), with an injection volume of 1 mL min−1 for 6 minutes. The peak was observed at 4.5 minutes.
 |
| | Fig. 1 Photocatalytic reactor set up for the degradation experiments. The set-up consists of (1) 300 W Hg lamp (2) glass tubes (50 mL) (3) air pump (4) water inlet (5) water outlet (6) air pump compressor (7) AC (8) top lid (9) bottom lid (10) fan (11) substrate for whole reactor (12) borosilicate condenser. | |
Similarly, in the case of AB-52, 50 mL of the dye solution containing an appropriate quantity of the catalyst suspensions was used. AB-52 solutions (25 mg L−1) containing 50 mg of photocatalyst samples were put in a sealed glass beaker and first ultrasonicated, and then stirred in the dark for 1 h to ensure absorption–desorption equilibrium. Afterwards it was irradiated by visible light in the reactor. 2 mL of samples were taken out at regular time intervals and separated through centrifugation. The supernatants were analyzed by recording variations in the absorption band maximum in the UV-Vis spectra of the dye molecule by using a JASCO V-670 UV-Vis spectrophotometer.
Analytical methods
HPLC-MS analysis for the EE2 degraded metabolites was carried out using a Thermo Finnigan Surveyor-Thermo LCQ Deca XP MAX with a BDS HYPERSIL C-18 (4.6 × 250 mm, 5.0 μm) HPLC column; while an Agilent 1260 series with an Eclipse XDBC18 (5 mm) reverse-phase column (4.6 × 150 mm) was used for the reverse-phase separation of the AB-52 degraded sample. The mobile phase was a mixture of methanol and water (40:60 v/v) at a flow rate of 0.2 mL min−1 for 60 min in the case of Acid Black-52, while a mixture of methanol and water at 30:70 (v/v) was used for EE2 for about 60 min. The mass spectra were obtained using Electro Spray Ionization (ESI) under a flow of helium gas at approximately 1 mL min−1 and the fragment voltage was 16 V.
Active species scavenging experiments
To identify the main active species involved in the photocatalytic reaction and degradation process, a series of scavenging experiments were conducted. Ascorbic acid as O2˙− scavenger,17 EDTA as h+ scavenger,18 potassium dichromate as e− scavenger,19 isopropanol as ˙OH scavenger,20 and potassium iodide (KI) as both ˙OH and h+ scavenger17,18 were used during the photocatalytic reaction experiments.
Active species quantification experiments
To investigate the main active species, hydroxyl radicals (˙OH) and superoxide radicals (˙O2−) produced during the photoreaction process, terepthalic acid (TA) and nitro blue tetrazolium (NBT) were utilized, respectively. To quantify the production of ˙OH radicals, 50 mg of the photocatalyst was added to a 50 mL mixture of TA solution (3 mmol) and NaOH (10 mmol). The production of ˙OH radicals under visible light irradiation was investigated by a HITACHI F-7000 Fluorescence Spectrophotometer. TA readily reacts with ˙OH hydroxyl radicals to generate 2-hydroxyterephthalic acid (TAOH) which emits fluorescence around 426 nm on excitation of its own 312 nm absorption band (Scheme 1). The increase in photoluminescent intensity of TAOH with time should be directly proportional to the generation of ˙OH radicals. Similarly, NBT (2 × 10−4 M) was used to detect the amount of ˙O2− radical generation from 50 mg of catalyst. The amount of ˙O2− generated during the photocatalytic reaction was monitored through the evolution of NBT, having an absorption maximum at 259 nm. NBT can be particularly reduced by photogenerated ˙O2− forming insoluble purple formazan compounds in aqueous solution (Scheme 2). The generation of ˙O2− was quantitatively analyzed by finding the decrease in NBT concentration in the supernatant solutions with a UV-Vis spectrophotometer (Thermo Scientific Orion aqua mate 8000).
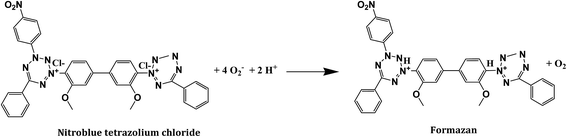 |
| | Scheme 1 Reaction pathway between NBT and superoxide radicals with the formation of formazan. | |
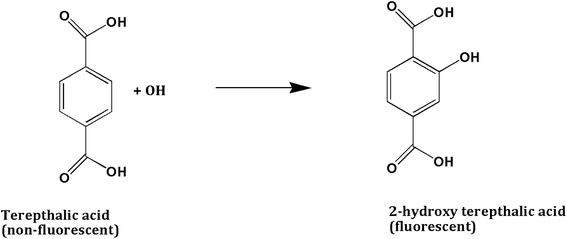 |
| | Scheme 2 Reaction pathway between terepthalic acid and hydroxyl radical with the formation of fluorescent 2-hydroxy terepthalic acid. | |
Phytotoxicity test
The phytotoxicity assessment test was carried out with seeds of V. radiata and P. vulgaris using a 100 ppm concentration of AB-52, EE2 and their corresponding degraded metabolites. The ethyl acetate extracted metabolites of AB-52 and EE2 were dried and dissolved in 10 mL of distilled water to give a final concentration of 100 ppm. The systematic methodology was based on the prescribed guidelines21,22 with few changes. The seeds (10 each) were sterilized in 10% mercuric chloride solution for 10 min and rinsed thoroughly with distilled water and used for interaction with dye, EE2 and corresponding degraded metabolites. The interaction was carried out in 100 mL Erlenmeyer flasks containing 100 ppm of pollutants and their respective degraded metabolites. Seeds in distilled water were included alongside as a control. Interacted seeds were placed on wet cotton in a Petri dish and incubated at 25 ± 1 °C in the dark for 24 h. Germinated seeds were selected for the toxicity study. The assay was carried out in standard beakers with 30 mL of 1.5% agar media (autoclaved) containing 100 ppm of AB-52, EE2 and degraded metabolites. The beakers with agar were immediately hardened in a freezer. The germinated seedlings were placed just above the surface of the agar in each beaker and incubated at 25 ± 1 °C in the dark. A control was included with seedlings on agar. After 5 days the plants were separated from the agar media and the root and shoot length was measured. All the experiments were carried out in triplicate and average data was produced in the present study.
Results and discussion
Characterization
UV-Vis absorption spectra (DRS mode) and photoluminescence spectra (PL) of the photocatalyst were analyzed in order to understand the mechanism involved in effective visible light absorption. UV-Vis absorption spectra of pure TiO2, Ag–TiO2 and Zr/Ag–TiO2 are shown in Fig. S1(a).† A well-defined band edge observed in the UV region of 300–350 nm could be ascribed to the photo-excitation occurring from the valence band to the conduction band. The extrapolation of a Kubelka Munk plot of hν vs. (αhν)2 was applied to get the optical energy band gap values (Eg), as shown in Fig. S1(b).† The figure shows that the energy band gap values for the pure TiO2, Ag–TiO2 and Zr/Ag–TiO2 nanoparticles were found to be approximately 3.18 eV, 3.08 eV and 2.87 eV, respectively. It was noticed that the doping of TiO2 with two transition metals (silver and zirconium) into its lattice could be accompanied with a decrease in the energy band gap and an increase in the wavelength (red shift). These results indicated that Zr/Ag–TiO2 nanoparticles have a greater possibility of exhibiting higher photo-catalytic activity in the visible region. Fig. S1(c)† shows the photoluminescence spectra of the prepared composites. The PL spectrum of the undoped TiO2 is also exhibited for comparison. All the spectra show near band gap emission (NBE) and blue or deep-level emission. The PL spectra suggest quelling of NBE on doping TiO2 with silver and zirconium. This may be due to the suppression of recombination of the photogenerated electron–hole pairs on doped TiO2.
Fig. 2 shows the X-ray diffraction pattern of pure TiO2, Ag–TiO2 and Zr/Ag–TiO2 nanoparticles. The main diffraction peaks observed in the patterns correspond to the TiO2 anatase crystalline phase (JCPDS 21-1272). The diffraction peaks corresponding to metallic zirconium or silver have not been observed, which suggests their doping did not affect the phase crystallinity. Furthermore, the represented oxide compounds (ZrxOy or AgxOy) are not observed either. This indicates that zirconium and silver nanoparticles are highly disseminated in the TiO2 crystal lattice. These doped metals did not show any phase changes, suggesting that aggregates might have been formed on the crystal borders and on the surface of the photocatalyst, thus elevating visible light absorption, as discussed earlier.
 |
| | Fig. 2 X-ray diffraction pattern of synthesized (a) Zr/Ag–TiO2 and (b) Ag–TiO2 nanoparticles. | |
Fig. S2(a) and S3(a),† show TEM images of Ag–TiO2 & Zr/Ag–TiO2 with uniform nano-crystalline structures and consistent grain size, as indicated by the XRD data. From the images, it could be understood that the uniformly distributed particles range from 10 to 17 nm and 7 to 15 nm, for Ag–TiO2 & Zr/Ag–TiO2, respectively. The SAED patterns of these samples are shown in Fig. S2(b) and S3(b),† in which the dark rings on the right correspond to the standard polycrystalline diffraction rings for the anatase phase (indexed). Evidently, no signals of diffraction rings associated with other phases were observed. The EDX profile [Fig. S2(d) and S3(d)†], revealed the composition of the elements present in the prepared samples. In the case of the Ag–TiO2 sample the weight% of Ag, Ti, and O was found to be 25.09, 24.73, and 50.18. Similarly, in Zr/Ag–TiO2 the weight% of Zr, Ag, Ti and O was found to be 20.08, 19.19, 19.87 and 40.86, respectively. The stoichiometry of both AgTiO2 (1:1:2) and ZrAgTiO2 (1:1:1:2) was confirmed by this measurement. HR-TEM images were [Fig. S2(c) and S3(c)†] used to obtain evident microstructure information in order to accurately analyze the single grains and grain boundaries.
In order to confirm the surface composition and chemical state of the Zr/Ag–TiO2 powders prepared (0.8 mole%), XPS analysis was carried out on a Kratos Axis Ultra 165 Spectrometer with Al Kα radiation. The discernible XPS scans were obtained for Ag 3d, Zr 3d, Ti 2p and O 1s to differentiate whether the dopant atoms were interwoven into the crystal lattice of TiO2 or formed different compounds with Ti, O, and dopant elements. Fig. 3(a) shows the XPS spectrum of Ag at 3d core levels. The Ag 3d5/2 and Ag 3d3/2 core level binding energies appearing at 368.1 and 374.1 eV, respectively, are in good agreement with bulk silver metallic values.23,24 The Zr 3d core level peaks observed at 182.4 and 184.6 eV corresponding to Zr 3d5/2 and Zr 3d3/2, respectively [Fig. 3(b)], are attributed to the +4 oxidation state of zirconium.25 The binding energies of Ti 2p [Fig. 3(c)] photoelectron peaks at 459.2 and 464.9 eV correspond to Ti 2p3/2 and Ti 2p1/2 lines, respectively.26,27 These values indicate that titanium is present in the +4 oxidation state. Fig. 3(d) shows the broad spectrum of the Zr/Ag–TiO2 nanocomposite. These results indicated that in the Ag/Zr–TiO2 nanocomposite, silver, titanium and zirconium metal ions are present in their highest oxidation state. The existence of Ag and Zr in the broad XPS spectrum and detection of no new compound between Ag, Zr, O, and Ti atoms other than TiO2, clearly indicate that Ag and Zr atoms are doped into the TiO2 crystal lattice.
 |
| | Fig. 3 XPS spectra of Ag 3d (a) Zr 3d (b) Ti 2p (c) and broad spectrum (d) of Zr/Ag–TiO2 nanocomposite. | |
The N2 adsorption–desorption isotherms and pore size distributions of the prepared materials are shown in Fig. 4. According to IUPAC, these isotherms are considered to be type IV, with characteristic TiO2 mesoporous materials. The specific surface areas of pure TiO2, Ag–TiO2 and Zr/Ag–TiO2 were observed to be 144.47, 147.83 and 151.44 m2 g−1, respectively. The pore size distributions (inset in Fig. 4) of the samples are about 14, 12 nm and 10 nm for TiO2, Ag–TiO2 and Zr/Ag–TiO2, respectively. The obtained results indicated that the surface area of the photocatalyst may slightly enhance the photo-activity, because Zr and Ag doped TiO2 with a large surface area has more active sites, so the organic molecules can be adsorbed in large quantities onto the TiO2 surface, and porosity facilitates pollutant access, adsorption, and decomposition.28,29 Based on these results, Zr/Ag–TiO2 was used for further photocatalytic tests.
 |
| | Fig. 4 N2 adsorption–desorption isotherms of as prepared composites (pore size distribution is the inset). | |
Photocatalytic degradation of EE2 and AB-52
To understand the photocatalytic activity of the Zr/Ag–TiO2 nanocomposite sample, its kinetic behaviour was investigated further using 17α-ethinyl estradiol (5 ppm) and Acid Black-52 (25 ppm). The photocatalytic degradation reaction kinetics of EE2 and AB-52 can be described by a Langmuir–Hinshelwood model. This indicated that the reactions occurred at a solid–liquid interface and the rate constant can be calculated from the following equation.| | |
ln[Co/Ct] = krKt = Kaprt
| (1) |
The plot of ln(Co/Ct) versus the irradiation time interval with various photocatalysts for 5 h under visible light irradiation is shown in Fig. 5. The results revealed that degradation of both EE2 (Fig. 5(a)) and AB-52 (Fig. 5(b)) follows first-order kinetics because the regression coefficients (R2) are all above 0.8856. Evidently, among the tested photocatalysts for degradation of EE2 and AB-52, the Zr/Ag–TiO2 nanocomposite (50 mg) exhibits the best performance, with rate constants k = 0.1617 min−1 and k = 0.1967 min−1, respectively (Table 1).
 |
| | Fig. 5 Degradation kinetics of EE2 (a) and AB-52 (b) under visible light irradiation at different time intervals. | |
Table 1 Rate constant (k) and linear correlation coefficient (R2) of the first-order kinetic reaction for EE2 and AB-52 degradation
| Sample |
EE2 |
AB-52 |
| k (min−1) |
R2 |
k (min−1) |
R2 |
| No catalyst |
0.0009 |
0.8351 |
0.0005 |
0.9233 |
| TiO2 alone |
0.0165 |
0.8981 |
0.0083 |
0.9541 |
| Ag–TiO2 |
0.0885 |
0.9914 |
0.1455 |
0.9690 |
| ZrAgTiO2 (25 mg) |
0.1482 |
0.9860 |
0.1601 |
0.9799 |
| ZrAgTiO2 (50 mg) |
0.1617 |
0.9882 |
0.1967 |
0.9918 |
To further investigate the possible intermediates and pathway during the degradation process by the Zr/Ag–TiO2 nanocomposite, LC-MS analysis was carried out for both EE2 and AB-52 samples. The degradation of EE2 and AB-52 was initially confirmed by HPLC analysis. The HPLC analysis report for both the samples is shown in Fig. S4 and S5.† At 0 h EE2 showed a single peak at a retention time of 1.54 min. After a period of 5 h as the photo degradation proceeded, the HPLC chromatogram of the degraded sample showed the presence of new peaks at 1.27 and 1.90 min, respectively. Similarly, at 0 h AB-52 showed a single dye peak at a retention time of 1.47 min. While the degradation proceeded, the sample showed new peaks at 1.42 and 1.54 min, respectively. All the HPLC profiles indicate the fragmentation of both the organic molecules into various compounds.
The LC-MS analysis of EE2 and AB-52 and their degradation products confirms the photocatalytic degradation of both organic samples by Zr/Ag–TiO2 nanoparticles under visible light irradiation after 5 h. LC-MS analysis of the AB-52 dye degraded sample demonstrated the presence of compounds with molecular weights of 327.3, 284.2 and 260.0, which could be interpreted as (M − 4), (M − 3) and (M+) peaks of structures A, B and C (Fig. 6 and S6†). LC-MS analysis of dye-degraded product also showed (M + 1), (M + 1), (M − 3), (M − 1), (M + 2), (M + 1), (M + 1) and (M + 1) peaks with molecular masses of 222.2, 190.2, 175.2, 316.4, 158.2, 134.1, 159.1 and 174.1 which represent the structures D, E, F, G, H, I, J and K (Fig. 6 and S6†). The degradation pathway indicates that the dye molecule has been degraded into various compounds after irradiation by visible light for 5 h.
 |
| | Fig. 6 Plausible degradation pattern of AB-52 under visible light irradiation using Zr/Ag–TiO2 (50 mg). | |
Similarly, the LC-MS pattern of the EE2 degraded sample showed the presence of compounds with molecular weights of 284.9, 238.1 and 222.3, which could be interpreted as (M + 3), (M + 2) and (M + 1) peaks of structures A, B and C (Fig. 7 and S7†). The degraded sample also showed (M − 2), (M + 2), (M − 1), (M + 2), (M − 3) and (M + 2) peaks with molecular masses of 214.2, 206.2, 178.2, 120.1, 103.1 and 82.2 which represents the structures D, E, F, G, H and I (Fig. 7 and S7†). The degradation pathway indicates that in 5 h of irradiation under visible light the EE2 molecule has been degraded into various compounds.
 |
| | Fig. 7 Plausible degradation pattern of EE2 under visible light irradiation using Zr/Ag–TiO2 (50 mg). | |
Phytotoxicity
The release of the untreated dyeing effluents and EDCs not only causes severe environmental and health hazards but also has a direct impact on the soil, affecting its fertility. Hence, it is mandatory to assess the phytotoxicity of the dye, EDC and degraded metabolites. The effects of AB-52, EE2 and their corresponding degraded metabolites at 100 ppm concentration on seeds of V. radiata and P. vulgaris were evaluated. The seeds treated with a 100 ppm concentration of AB-52 showed low germination percentage in V. radiata (30%), and P. vulgaris (40%). Similarly, seeds treated with EE2 also showed less germination in V. radiata (40%), and P. vulgaris (50%). In contrast, seeds treated with a 100 ppm concentration of degraded metabolites and distilled water exhibited a 100% germination rate in both seeds. The root and shoot length were found to be significantly affected by AB-52 and EE2 compared to their corresponding metabolites after degradation and with distilled water (Table 2) (Fig. 8). The results of the phytotoxicity study indicate the ability of the co-doped nanoparticles to not only degrade AB-52 and EE2 but also to detoxify it.
Table 2 Phytotoxicity assessment of AB-52, EE2 and their corresponding extracted intermediates
| |
Vigna radiata |
Phaseolus vulgaris |
| Germination (%) |
Shoot (cm) |
Root (cm) |
Germination (%) |
Shoot (cm) |
Root (cm) |
| Water |
100 |
10.27 ± 0.57 |
4.67 ± 0.63 |
100 |
6.65 ± 1.31 |
2.47 ± 0.54 |
| AB-52 |
30 |
2.12 ± 0.15 |
0.43 ± 0.15 |
30 |
2.76 ± 0.76 |
0.66 ± 0.15 |
| AB-52 metabolites |
100 |
6.45 ± 1.21 |
1.15 ± 0.24 |
100 |
4.45 ± 0.42 |
1.31 ± 0.25 |
| EE2 |
40 |
3.30 ± 0.31 |
0.72 ± 0.17 |
50 |
3.21 ± 0.83 |
0.92 ± 0.17 |
| EE2 metabolites |
100 |
5.15 ± 0.51 |
1.92 ± 0.24 |
100 |
4.65 ± 0.40 |
1.43 ± 0.25 |
 |
| | Fig. 8 Phytotoxicity assessment of AB-52, EE2 and their corresponding metabolites. | |
Active species scavenging experiment
Different scavenging experiments have been carried out to investigate the main active species for the photocatalytic degradation process of both EE2 and AB-52. Ascorbic acid was used as O2˙− scavenger, EDTA as h+ scavenger, K2Cr2O7 as e− scavenger, isopropylalcohol as ˙OH scavenger, and potassium iodide (KI) was used as both ˙OH and h+ scavenger (Fig. 9(a) and (b)). After the addition of the scavengers to each reaction solution, the photocatalytic activity decreased in the order isopropanol > ascorbic acid > EDTA > potassium iodide > potassium dichromate > no scavenger. These results suggested that intervention in the degradation of EE2 and AB-52 was greater with the addition of isopropyl alcohol and ascorbic acid, which shows that ˙OH and O2˙− are the primary species in the photocatalytic degradation process. Further, the addition of EDTA which scavenges h+ and K2Cr2O7 as an e− scavenger showed no significant decrease in the photocatalytic degradation process, implying that the hydroxyl and superoxide radicals are the primary species responsible for an efficient degradation process.
 |
| | Fig. 9 Active species scavenging experiments using Zr/Ag–TiO2 during EE2 (a) and AB-52 (b) degradation. | |
Active species quantification
Further to quantify ˙OH and O2˙− radicals, probing techniques have been carried out using NBT and TA. Quantification experiments of ˙O2− production have been done through the transformation of NBT (a detection agent for ˙O2−) during the photocatalytic reaction30 (Scheme 1). Fig. 10(d) shows the kinetic transformation percentage of NBT at different time intervals. It could be seen that Zr/Ag–TiO2 nanoparticles showed a higher transformation percentage of NBT than TiO2, indicating that the generation of ˙O2− increased with an increase in time [Fig. 10(c)]. This could be attributed to the lower band gap energy of Zr/Ag–TiO2 (2.87 eV), as shown in a Tauc plot (Fig. S1(b)†), and also to the lower recombination rate of the photogenerated electron–hole pairs on doping, as described by PL intensities (Fig. S1(c)†). During the photoexcitation of the catalyst, most of the electrons generated remain in the CB (conduction band) to react with O2 to generate more ˙O2−. This was proved by the highest transformation concentration for NBT over Zr/Ag–TiO2, as shown in Fig. 10(d). Due to a smaller band gap, the photogenerated electrons in the VB (valence band) can easily transfer to the CB of Zr/Ag–TiO2, resulting in more effective charge separation and reducing the recombination of electron–hole pairs, which finally leads to more photogenerated electrons for reaction with O2 to produce ˙O2− which can take part in decomposition of EE2 and AB-52, which is in good agreement with the active scavenging experiments (Fig. 9).
 |
| | Fig. 10 Concentration of ˙OH (a) and ˙O2− (c) radicals; formation of TAOH (b) and NBT transformation to formazan products (d) at different time intervals. | |
To further quantify ˙OH radicals, the terephthalic acid photoluminescence probing technique (TA-PL) was employed, since TA could readily react with ˙OH radicals to form a highly fluorescent 2-hydroxyterephthalic acid (TAOH)31–33 (Scheme 2). As expected, the TAOH concentration over Zr/Ag–TiO2 nanoparticles increased with an increase in time compared to other prepared nanoparticles [Fig. 10(b)]. This could be attributed to the easy charge transfer, which results in an improvement in the number of holes on the VB of Zr/Ag–TiO2 to produce more ˙OH, resulting in the highest PL peak of Zr/Ag–TiO2 with respect to time. Fig. 10(a) shows the concentration of ˙OH radicals with respect to time. Based on the results of the NBT transformation and the TA-PL results, it can be concluded that ˙O2− and ˙OH radicals play a major role in the photodegradation of both the pollutants EE2 and AB-52 over Zr/Ag–TiO2 nanoparticles and that there is a linear correlation with the concentration of reactive organic species during visible light photodegradation.
Mechanism of degradation
The possible photocatalytic mechanism was used to understand the enhanced visible activity of the as-prepared Zr/Ag–TiO2 catalyst. After doping of Zr and Ag, the band gap of TiO2 was narrowed from 2.87 eV. Thus, the visible light can be absorbed by Zr/Ag–TiO2 catalysts with improved efficiency. The electrons in the valence band of Zr/Ag–TiO2 could be excited to the conduction band with ease and a large amount of electron–hole pairs is then generated. It is well demonstrated that ‘Ag’ traps the electrons from the CB of TiO2. The doping of ‘Zr’ also suppresses the electron–hole recombination by electron trapping.34 Subsequently, with a suitable doping concentration, the doped Zr and Ag species in the lattice structure of TiO2 served to ameliorate the electronic conductivity, which significantly suppressed the recombination of photogenerated electron–hole pairs and led to higher energy efficiency.
During the photodegradation process, the electrons from the valance band of Zr/Ag–TiO2 can migrate easily to the conduction band by absorbing visible light, leaving holes in the valence band (eqn (2)). These photogenerated electrons from the conduction band are scavenged by surrounding oxygen absorbed on the surface of the product, converting the radicals into superoxide anion radicals (˙O2−), which are responsible for the photodegradation of both the organic pollutants (eqn (3)). Similarly, the photogenerated holes in the VB of TiO2 readily oxidize H2O and OH− to form ˙OH radicals (eqn (4) and (5)), which are further involved in the photodegradation of EE2 and AB-52. Finally, the formed hydroxyl (˙OH) and superoxide (˙O2−), radicals are highly reactive oxidizing species that will react with the EE2 and AB-52 on the catalyst surface to form the degradation products (eqn (6)), the proposed mechanism is expressed as follows:
| | |
Zr/Ag–TiO2 + hν → Zr/Ag–TiO2 (eCB− + hVB+)
| (2) |
| | |
Zr/Ag–TiO2 (eCB−) + O2 → Zr/Ag–TiO2 + ˙O2
| (3) |
| | |
Zr/Ag–TiO2 (hVB+) + H2O → Zr/Ag–TiO2 + ˙OH + H+
| (4) |
| | |
Zr/Ag–TiO2 (hVB+) + OH− → Zr/Ag–TiO2 + ˙OH
| (5) |
| | |
EE2 or AB-52 + ˙O2− + ˙OH → degradation product
| (6) |
Conclusion
TiO2 nanoparticles co-doped with silver and zirconium were prepared for effective utilization in degrading two toxic organic pollutants: 17α-ethinyl estradiol and Acid Black-52. A simple multi-tube photoreactor set-up was developed and utilized for degradation experiments. Recombination of photogenerated electron–hole pairs has been significantly suppressed by doping impurities into the TiO2 crystal lattice. The degradation pathway and possible intermediate compounds during degradation were investigated by LC-MS analysis. In order to evaluate the phytotoxicity of intermediate products during the photodegradation process, two different seeds V. radiata and P. vulgaris were utilized. The toxicity analysis suggested that degraded metabolites showed a less toxic nature compared to normal EE2 and AB-52. We believe that the present work demonstrates the ability of multifunctional nanomaterials not only to degrade the hazardous pollutants but also to detoxify them during photo catalysis.
Acknowledgements
The study was financially supported by the National Natural Science Foundation of China for Foreign Youth Scholars (Grant No. 51650110501), the China Postdoctoral Science Foundation (Grant No. 2016M591767), the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (Grant No. 51421006), Program for Environmental Protection in Jiangsu Province (Grant No. 2015037), and Priority Academic Program Development of Jiangsu Higher Education Institutions. The Fundamental Research Funds for the Central Universities (Grant No. 2016B10614), Priority Academic Program Development of Jiangsu Higher Education Institutions, and Top-notch Academic Programs project of Jiangsu Higher Education Institutions (TAPP).
References
- M. S. U. Rehman, N. Rashid, M. Ashfaq, A. Saif, N. Ahmad and J. I. Han, Chemosphere, 2015, 138, 1045–1055 CrossRef CAS PubMed.
- P. Martínez, L. M. Faria, J. L. Do-na-Rodríguez, J. M. Fernández-Rodríguez and C. Silva, Appl. Catal., B, 2012, 113–114, 221–227 CrossRef.
- R. Mohammadi, B. Massoumi and M. Rabani, Int. J. Photoenergy, 2012, 1–11 CrossRef.
- T. Deblonde, C. Cossu-Leguille and P. Hartemann, Int. J. Hyg. Environ. Health, 2011, 214, 442–448 CrossRef CAS PubMed.
- D. L. Lima, V. Calisto and V. I. Esteves, Chemosphere, 2011, 84, 1072–1078 CrossRef CAS PubMed.
- D. Nasuhoglu, D. Berk and V. Yargeau, Chem. Eng. J., 2012, 185, 52–60 CrossRef.
- A. Kumar and I. Xagoraraki, Sci. Total Environ., 2010, 408, 5972–5989 CrossRef CAS PubMed.
- S. Hwang, D. I. Lee, C. H. Lee and I. S. Ahn, J. Hazard. Mater., 2008, 155, 334–341 CrossRef CAS PubMed.
- Z. Zhang, Y. Feng, Y. Liu, Q. Sun, P. Gao and N. Ren, J. Hazard. Mater., 2010, 181, 1127–1133 CrossRef CAS PubMed.
- C. E. J. R. Desbrow, E. J. Routledge, G. C. Brighty, J. P. Sumpter and M. Waldock, Environ. Sci. Technol., 1998, 32, 1549–1558 CrossRef CAS.
- G. R. Li, D. L. Qu, W. X. Zhao and Y. X. Tong, Electrochem. Commun., 2007, 9, 1661–1666 CrossRef CAS.
- H. Serier, O. Toulemonde, D. Bernard, A. Demourgues, J. Majimel and M. Gaudon, Mater. Res. Bull., 2012, 47, 755–762 CrossRef CAS.
- Z. Wang, C. Chen, F. Wu, B. Zou, M. Zhao, J. Wang and C. Feng, J. Hazard. Mater., 2009, 164, 615–620 CrossRef CAS PubMed.
- A. Hernández-Gordillo and V. Rodríguez González, Chem. Eng. J., 2015, 261, 53–59 CrossRef.
- Q. Chen, Y. Xin and X. Zhu, Electrochim. Acta, 2015, 186, 34–42 CrossRef CAS.
- Y. Yoon, P. Westerhoff, S. A. Snyder and M. Esparza, Water Res., 2003, 37, 3530–3537 CrossRef CAS PubMed.
- S. Song, L. Xu, Z. He, H. Ying, J. Chen, X. Xi and B. Yan, J. Hazard. Mater., 2008, 152, 1301–1308 CrossRef CAS PubMed.
- Y. Fu, H. Chen, X. Sun and X. Wang, Appl. Catal., B, 2012, 111–112, 280–287 CrossRef CAS.
- L. S. Zhang, K. H. Wong, H. Y. Yip, C. Hu, J. C. Yu, C. Y. Chan and P. K. Wong, Environ. Sci. Technol., 2010, 44, 1392–1398 CrossRef CAS PubMed.
- Z. Pan, E. Stemmler, H. J. Cho, W. Fan, L. LeBlanc, H. H. Patterson and A. Amirbahman, J. Hazard. Mater., 2014, 279, 17–25 CrossRef CAS PubMed.
- USEPA, Ecological effects test guidelines. Seed germination root elongationtoxicity test, Office of prevention, Pesticides and Toxic substances 850 4200, Washington DC, EPA 712-C-96-163, 1996 Search PubMed.
- A. S. Arun Prasad, V. S. V. Satyanarayana and K. V. Bhaskar Rao, J. Hazard. Mater., 2013, 262, 674–684 CrossRef CAS PubMed.
- E. Sumesh, M. S. Bootharaju and A. T. Pradeep, J. Hazard. Mater., 2011, 189, 450–457 CrossRef CAS PubMed.
- Y. Lai, H. Zhang, K. Xie, D. Gong, Y. Tang, L. Sun, C. Lin and Z. Chen, New J. Chem., 2010, 34, 1335–1340 RSC.
- T. sunekawa, K. Asami, S. Ito, M. Yashima and T. Sugimoto, Appl. Surf. Sci., 2005, 252, 1651–1656 CrossRef.
- K. V. Bineesh, S. Y. Kim, B. R. Jermy and D. W. Park, J. Ind. Eng. Chem., 2009, 15, 207–211 CrossRef CAS.
- Y. Kim, J. Lee, H. Jeong, Y. Lee, M. H. Um, K. M. Jeong, M. K. Yeo and M. Kang, J. Ind. Eng. Chem., 2008, 14, 396–400 CrossRef CAS.
- A. Zielińska-Jurek and A. Zaleska, Catal. Today, 2014, 230, 104–111 CrossRef.
- Y. L. Shang, Y. L. Jia, F. H. Liao, J. R. Li, M. X. Li, J. Wang and S. H. Zhang, J. Mater. Sci., 2007, 42, 2586–2590 CrossRef CAS.
- K. I. Ishibashi, A. Fujishima, T. Watanab and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- N. Arunkumar and R. Vijayaraghavan, RSC Adv., 2014, 4, 65223–65231 RSC.
- S. Naraginti, Y. Li and Y. Wu, RSC Adv., 2016, 6, 75724–75735 RSC.
- N. Tian, H. Huang, Y. He, Y. Guo, T. Zhang and Y. Zhang, Dalton Trans., 2015, 7, 4297–4307 RSC.
- S. T. Pantelides, Rev. Mod. Phys., 1978, 50, 797–858 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20702b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 











