
An efficient and rapid intramolecular cyclization of a quadruple Mannich reaction for one-pot synthesis of pentaazaphenalenes and their antimicrobial activities†
Ahmed F. M. EL-Mahdy* and
Hassan A. H. EL-Sherief
Chemistry Department, Faculty of Science, Assiut University, Assiut 71516, Egypt. E-mail: ahmed.ahmed20@science.au.edu.eg; Fax: +20 882080209; Tel: +20 1007799743
First published on 21st September 2016
Abstract
A simple, rapid and one-pot quadruple Mannich reaction has been developed for the synthesis of 2,5,7,9,11-pentaazaphenalenes via a three-component reaction of 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one, formaldehyde and primary amines in ethanol at room temperature. The reaction products were obtained in very good to excellent yields using a simple work-up procedure. The regioselectivity of the reaction was studied using the electronic energy calculations which indicated that the corresponding products 2,5,7,9,11-pentaazaphenalenes are more favorable than the other isomeric products 3,5,8,10,11-pentaazaanthracenes and 2,5,7,8,10-pentazaphenanthrenes. The reaction mechanism was investigated and the structures of all the new compounds were confirmed using spectra and elemental analysis. In addition, all the synthesized compounds were screened for antimicrobial activity and showed a significant activity against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Candida albicans, Geotrichum candidum and Trichophyton rubrum.
1. Introduction
Discovering new bioactive compounds with a minimum number of synthetic steps and in much less time is a great challenge to chemists.1 Previously, multi-step reactions which were associated with low yield, large amount of waste, high cost, difficulty in product purification and isolation, have been frequently used to prepare bioactive compounds. Recently, multicomponent reactions (MCRs) in which three or more components are combined together in one synthetic operation have gained considerable and steadily increasing interest.2 In comparison with the multi-step reactions, the significant attribution of MCRs is the formation of several bonds in one synthetic operation without changing the reaction conditions, isolating the intermediates or adding further reagents. Thus, avoiding the complicated purification operations and saving of both solvents and reagents.3 Therefore, MCRs have served as a powerful tool in drug discovery, medicinal chemistry4 and pharmaceutical industry.5Pyrimidines have received much attention in medicinal chemistry due to their biological activities and therapeutic applications. One possible reason for their biological activities is the presence of pyrimidine base as nucleobases (uracil, thymine and cytosine, Fig. 1), which are essential building blocks of nucleic acids, DNA and RNA. Compounds with pyrimidine ring system are widely used as antifungal,6 antibacterial,7 anti-inflammatory,8 anticancer,9 anti-convulsant,10 sedative,11 analgesic,12 anti-depressive13 and anti-pyretic agents.14 Pyrimidine as a heterocyclic nucleus is present in many modern drugs including stavudine (antiretroviral),15 lamivudine (anti-AIDS),16 brivudine (anticancer),17 nelarabine (antileukemia)18 and sulfadimethoxine (sulfonamide antibiotic) (Fig. 1). In addition, fused pyrimidines have a diverse pharmacological activity such as antitumor,19 anticancer,20 antihypertensive,21 antifolate22 and antioxidant.23 Due to the significant biological activity of pyrimidine derivatives, the synthesis of this class of compounds plays an attractive scaffold in the medicinal chemistry and drug discovering.
 | ||
| Fig. 1 Nucleobases and some drugs containing pyrimidine nucleus. | ||
Mannich reaction is a multicomponent and enormously useful reaction for the construction of nitrogenous molecules and the formation of C–C and C–N bonds.24 In the Mannich reaction, the aminoalkylation of compounds which have at least one active hydrogen atom, is performed through the condensation with formaldehyde (or another aldehyde) and ammonia (or ammonia derivatives). Additionally, double Mannich reaction can occur, if the starting compound contains two adjacent active hydrogen atoms.25–28 Recently, we have reported the utility of double Mannich reaction for the synthesis of pyrimidothiadiazines,26 triazolothiadiazines,27 pyrazolopyrimidines27a and thiadiazinobenzimidazoles28 in high yields. Previously, it has been reported that the double Mannich reaction of 6-amino-2-thiouracil or its N-1-derivatives with formaldehyde and primary aromatic amines in methanol containing acetic acid or in ethanol, afforded the corresponding pyrimido[4,5-d]pyrimidin-4-ones (Fig. 2a) or its N-1-derivatives (Fig. 2b), respectively.29,30
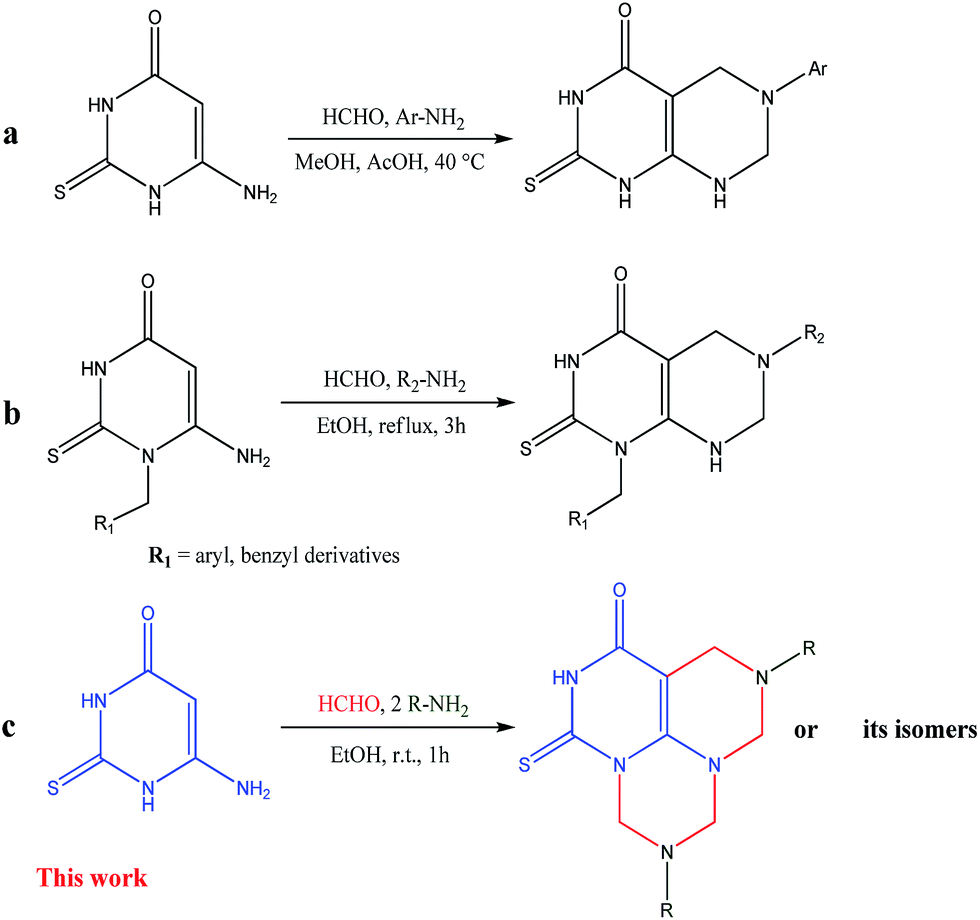 | ||
| Fig. 2 Strategies for Mannich reaction of 6-amino-2-thiouracil. | ||
As a part of our ongoing interest in the synthesis of diversely functionalized compounds of biological importance using Mannich type reaction, we report herein a simple, rapid and efficient quadruple Mannich reaction of 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (1) with formaldehyde and aromatic or aliphatic primary amines (2) in ethanol at room temperature for 1 h and then study the expected tricyclic products (Fig. 2c).
2. Results and discussion
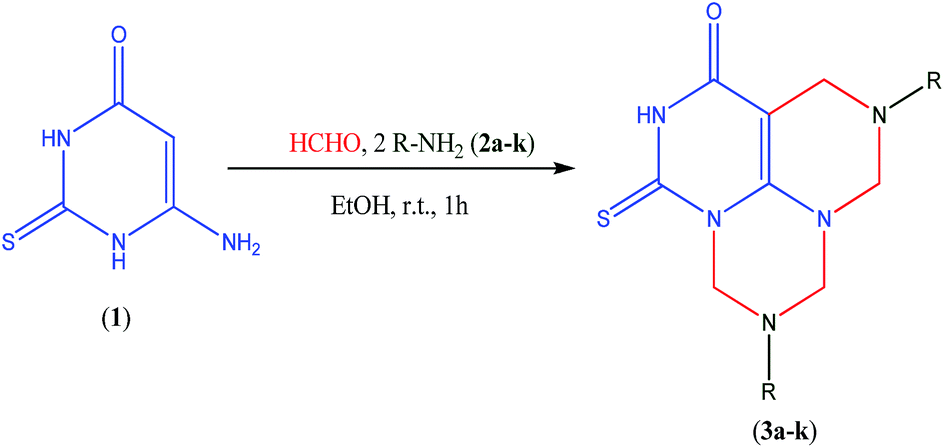
The objective of the present work is the investigation of the behaviour of quadruple Mannich reaction towards 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (1) as a polyfunctional nucleophile. On treatment of compound (1) with an excess of formaldehyde and two moles of aliphatic or aromatic amines (2a–k) in ethanol at room temperature for 1 h, the reaction may proceed to afford 5,9-disubstituted-1-thioxo-5,6,9,10-tetrahydro-4H,8H-2,5,7,9,11-pentaazaphenalene-3-ones (3a–k) or other possible isomeric products 3,8-disubstituted-3,4,8,9-tetrahydro-2H,7H-1-thia-3,5,8,10,11-pentaazaanthracene-6-ones (4a–k) and 5,10-disubstituted-5,6,10,11-tetrahydro-4H,9H-1-thia-2,5,7,8,10-pentazaphenanthrene-3-ones (5a–k) or a mixture of them. However, based on TLC, the reaction gave only a single product (Scheme 1). | ||
| Scheme 1 Quadruple Mannich reaction of 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (1). | ||
The IR spectra of the reaction products lacked the NH2 absorption peaks and showed characteristic bands at 3300–3451, 2893–2974, 1613–1679, 1588–1636 and 1489–1589 cm−1 attributed to NH, CH aliphatic, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN and CS, respectively. The 1H-NMR and 13C-NMR of the isolated products have confirmed the reaction of two molecules of amines and the performing of the quadruple Mannich reaction. Since, the 1H-NMR spectra of the products were characterized by the appearance of four singlet signals at δ = 3.45–4.11, 4.03–4.90, 4.16–5.10 and 5.15–6.35 ppm attributed to four methylene (CH2) groups, in addition to the other protons at the expected chemical shifts. While, the 13C-NMR spectra of the isolated products showed the four signals for the methylene (CH2) at δ = 64.41–67.69, 63.60–66.96, 63.15–64.08 and 45.13–58.25 ppm. Additionally, the mass spectra of the synthesized products showed the expected molecular ion peaks and their elemental analysis were found to be in agreement with the calculated ones. All these data can't confirm which one of these isomers 3, 4 and 5 is formed.
O, CN and CS, respectively. The 1H-NMR and 13C-NMR of the isolated products have confirmed the reaction of two molecules of amines and the performing of the quadruple Mannich reaction. Since, the 1H-NMR spectra of the products were characterized by the appearance of four singlet signals at δ = 3.45–4.11, 4.03–4.90, 4.16–5.10 and 5.15–6.35 ppm attributed to four methylene (CH2) groups, in addition to the other protons at the expected chemical shifts. While, the 13C-NMR spectra of the isolated products showed the four signals for the methylene (CH2) at δ = 64.41–67.69, 63.60–66.96, 63.15–64.08 and 45.13–58.25 ppm. Additionally, the mass spectra of the synthesized products showed the expected molecular ion peaks and their elemental analysis were found to be in agreement with the calculated ones. All these data can't confirm which one of these isomers 3, 4 and 5 is formed.
Previously, the double Mannich reaction of 6-amino-2-thiouracil (1) with formaldehyde and primary amines has been performed on active CH and NH2 groups to give the corresponding pyrimido[4,5-d]pyrimidin-4-ones (6a–k)29 (Fig. 2a). This reported result directed us to think about the tautomerism of the resulting compound, whether its structure existed in form 6a–k, 7a–k or 8a–k (Table 1). Based on the electronic energy calculations for the tautomers using Gaussian 09 (Table 1), we can conclude that the tautomer 6a–k is the most stable ones, which could be easily transformed to the final isomeric products 3a–k via a second double Mannich reaction (Scheme 2).
|
||||||
|---|---|---|---|---|---|---|
| R | Energy (a.u.) | E7a–k–E6a–kb (kcal mol−1) | E8a–k–E6a–kb (kcal mol−1) | |||
| 6a–ka | 7a–ka | 8a–ka | ||||
| a Numbers represent the electronic energy values in (a.u.) unit.b Numbers represent the difference values of electronic energies in (kcal mol−1) unit. “1 a.u. of energy = 627.5 kcal mol−1”. | ||||||
| a | CH2CH3 | −1004.56566593 | −1004.55224449 | −1004.52839361 | 8.42 | 23.39 |
| b | CH2CH(CH3)2 | −1083.19682552 | −1083.18363265 | −1083.15984570 | 8.28 | 23.21 |
| c | CH2CH2CH2CH3 | −1083.20012912 | −1083.18662357 | −1083.16279485 | 8.47 | 23.43 |
| d | C6H5 | −1156.99669096 | −1156.98402381 | −1156.96015799 | 7.95 | 22.92 |
| e | CH2C6H5 | −1196.31028377 | −1196.29684344 | −1196.27334491 | 8.43 | 23.18 |
| f | C6H4OCH3-p | −1271.52405724 | −1271.51105934 | −1271.48734032 | 8.16 | 23.04 |
| g | C6H4CH3-m | −1196.31779247 | −1196.30495082 | −1196.28118405 | 8.06 | 22.97 |
| h | C6H4CH3-p | −1196.31722738 | −1196.30435518 | −1196.28054666 | 8.08 | 23.02 |
| i | C6H4Cl-p | −1616.59021569 | −1616.57806458 | −1616.55402664 | 7.62 | 22.71 |
| j | C6H4Br-p | −3728.12332894 | −3728.11115241 | −3728.08708152 | 7.64 | 22.75 |
| k | 2-Naphthylamine | −1310.64677190 | −1310.63594291 | −1310.61201867 | 6.80 | 21.81 |

 | ||
| Scheme 2 Plausible mechanism for the formation of compounds 3a–k. | ||
The regioselectivity of the second intramolecular cyclization step is assumed to be the difference in the electron density at the N-1, N-3 and N-8 atoms of the intermediates 6a–k. Based on atomic charge calculations of 6a, the highest negative charge is located on N-8 (−0.67056) and the other N-1 and N-3 atoms have less negative charge of −0.61320 and −0.64060, respectively (see Fig. S1 and Table S1 in ESI†). Consequently, the second intramolecular cyclization step should be started from the most basic N-8 atom followed by cyclization with the near N-1 atom to afford the isomeric products 3a–k (Scheme 2). In addition, this assumption was supported by the electronic energy calculations for the expected products, which confirmed that the isomeric products 3a–k are more favorable and stable than the other expected isomers 4a–k and 5a–k (Table 2). Further, the obtained products were found to be easily dissolved in alcoholic KOH solution (5%) and reprecipitated again by addition of diluted acetic acid, which chemically in agreement with products 3a–k. Furthermore, the S-alkylated derivative of 6-amino-2-thiouracil undergoes only a triple Mannich reaction involving N-8 when treated with excess of formaldehyde and amines (unpublished data).
| R | Energy (a.u.) | E4a–k–E3a–kb (kcal mol−1) | E5a–k–E3a–kb (kcal mol−1) | |||
|---|---|---|---|---|---|---|
| 3a–ka | 4a–ka | 5a–ka | ||||
| a Numbers represent the electronic energy values in (a.u.) unit.b Numbers represent the difference values of electronic energies in (kcal mol−1) unit. 1 a.u. of energy = 627.5 kcal mol−1. | ||||||
| a | CH2CH3 | −1215.95190106 | −1215.94411353 | −1215.91174915 | 4.89 | 25.20 |
| b | CH2CH(CH3)2 | −1373.21456031 | −1373.21427457 | −1373.18190016 | 0.18 | 20.49 |
| c | CH2CH2CH2CH3 | −1373.22099819 | −1373.21301295 | −1373.18068271 | 5.01 | 25.30 |
| d | C6H5 | −1520.81626569 | −1520.81284994 | −1520.78057224 | 2.14 | 22.40 |
| e | CH2C6H5 | −1599.44174383 | −1599.43420956 | −1599.40252236 | 4.73 | 24.61 |
| f | C6H4OCH3-p | −1749.87115741 | −1749.86749079 | −1749.83616641 | 2.30 | 21.96 |
| g | C6H4CH3-m | −1599.45852274 | −1599.45488592 | −1599.42290102 | 2.28 | 22.35 |
| h | C6H4CH3-p | −1599.45742388 | −1599.45375149 | −1599.42190213 | 2.30 | 22.29 |
| i | C6H4Cl-p | −2440.00307987 | −2440.00037626 | −2439.96734212 | 1.70 | 22.43 |
| j | C6H4Br-p | −6663.06927705 | −6663.06637527 | −6663.03353198 | 1.82 | 22.43 |
| k | b-Naphthylamine | −1828.11938851 | −1828.11637803 | −1828.08274758 | 1.89 | 22.99 |
Formation of the isomeric products 3a–k was further evidenced by using 2D ROESY and HMBC NMR methods. The ROESY spectrum of product 3j as an example (see page S31 in ESI†) showed strong diagonal peaks at δ = 4.11, 4.84, 5.02 and 5.82 ppm attributed to the four methylene (CH2) groups, in addition to aromatic peaks at δ = 6.85 and 7.36 ppm. The isomeric product 3j did not show strong cross-peaks. A much weaker through-space correlations appeared between H4–H2′, H6–H6′, H8–H2′′ and H10–H6′′. It is very interesting to note that there is no cross-correlation of NH group with methylene groups. On the other hand, the 1H–13C HMBC spectrum of 3j was characterized by several cross-peaks of the four methylene groups (Fig. 3a). The methylene group (C-4) at δH = 4.11 ppm showed heteronuclear 3JH–C coupling with C-6, C-1′ and CO and 2JH–C coupling with C-3a. The methylene group (C-6) at δH = 4.84 ppm exhibited three 3JH–C coupling with C-4, C-8 and C-11a. In addition, the methylene group (C-8) at δH = 5.02 ppm showed four 3JH–C coupling with C-6, C-10, C-1′′ and C-11a. The last methylene group (C-10) at δH = 6.85 ppm showed four 3JH–C coupling with C-8, C-1′′, C-11a and CS. Furthermore, there is no evident correlation between the N–H at δH = 12.07 ppm and any of the four methylene groups, the only correlation coupling of N–H with C-3a. These 2D NMR data significantly supported the existence of the isomeric product in form 3j (Fig. 3b) not 4j or 5j.
 | ||
| Fig. 3 (A) HMBC spectrum of compound 3j in DMSO-d6. (B) Optimized minimum structure of compound 3j at the B3LYP/6-31+G(d,p) level. Atom color code: carbon (gray), nitrogen (blue), oxygen (red), sulfur (yellow), bromine (dark red) and hydrogen (white). | ||
The generality of the reaction was investigated by employing 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (1) with formaldehyde and various primary amines 2a–k in ethanol at room temperature for 1 h to produce the desired products 3a–k in very good to excellent yields (Table 3). It was observed that aliphatic amines such as ethylamine, isobutylamine and butylamines gave the corresponding products in excellent yields (Table 3, entries 1–3). Benzylamine gave a very good yield of the product (Table 3, entry 4). Aromatic amines are less active than aliphatic amines towards Mannich reaction. Therefore, aniline and 2-napthylamine produced the desired products in very good yields (Table 3, entries 5, 11). Additionally, aromatic amines with electron-donating substituents such as p-methoxyaniline, m-methylaniline and p-methylaniline, and that with electron-withdrawing substituents such as p-chloroaniline and p-bromoaniline led to very good to excellent yields (Table 3, entries 6–10). As previously reported, the Mannich reaction of uracil with formaldehyde and aromatic amine containing strong electron-withdrawing group such as p-nitroaniline has been failed to perform.31 On the same manner, the quadruple Mannich reaction of starting compound (1) with p-nitroaniline was examined and no product was obtained (Table 3, entries 12). It can be noticed that water is the sole by-product in the reaction process, which makes the reaction work-up very easy and convenient. In all cases, the obtained solid product was formed during stirring which was collected simply by filtration and then purified by recrystallization from the proper solvent.
3. Plausible mechanism
On the basis of experimental results and electronic energy calculations, a reasonable reaction mechanism of the formed compounds 3a–k is postulated in Scheme 2. With the first double Mannich reaction, the condensed imines which forms from the reaction of formaldehyde with primary amines, undergoes nucleophilic addition to the active CH group of compound (1) to produce the uncyclized intermediates 9a–k. The formed intermediates 9a–k then nucleophilic attacks another molecule of formaldehyde followed by intramolecular cyclization in the presence of an acid catalyst with the elimination of water molecule to produce the cyclized intermediates 6a–k. By the same manner, the second double Mannich reaction is performed on N-1 and N-8 of intermediates 6a–k to afford the desired products 3a–k.4. Antimicrobial activity
One of the important proposes of the present work is the synthesis of new heterocyclic compounds which might be of interest in biological and medicinal chemistry. Accordingly, all synthesized compounds were screened in vitro for their antimicrobial activity against three strains of bacteria (Escherichia coli, Pseudomonas aeruginosa and Staphylococcus aureus) and three strains of fungi (Candida albicans, Geotrichum candidum and Trichophyton rubrum) (Table 4) (Fig. 4). The inhibition zone (mm) and MIC (minimum inhibition concentration) of the screened compounds were compared with levofloxacin and clotrimazole which were used as a reference for antibacterial and antifungal tests, respectively.| Compound | Escherichia coli (−ve) | Pseudomonas aeruginosa (−ve) | Staphylococcus aureus (+ve) | Candida albicans | Geotrichum candidum | Trichophyton rubrum |
|---|---|---|---|---|---|---|
| a Numbers out parentheses represent the diamer of inhibition zone in (mm) of compounds 3a–k.b Numbers in parentheses represent the MIC (minimum inhibition concentration) in (μg mL−1) of compounds 3a–k. | ||||||
| 3a | 8a (17)b | 10 (20) | 9 (5.0) | — | 19 (8.0) | 19 (18) |
| 3b | — | 12 (15) | 11 (5.0) | — | 20 (8.0) | 20 (18) |
| 3c | 9 (16) | 11(15) | 13 (8.0) | 10 (15) | 22 (10) | 20 (16) |
| 3d | 13 (15) | 10 (10) | 12 (3.0) | 12 (12) | 22 (5.0) | 22 (12) |
| 3e | 14 (9.0) | 17 (10) | 13 (2.5) | 14 (10) | 25 (4.0) | 24 (16) |
| 3f | 16 (8.0) | 18 (12) | 13 (3.0) | 13 (10) | 23 (5.0) | 23 (12) |
| 3g | 13 (5.0) | 17 (10) | 12 (3.0) | 10 (12) | 23 (5.0) | 23 (12) |
| 3h | 12 (5.0) | 17 (10) | 12 (2.5) | 10 (6.0) | 22 (8.0) | 20 (12) |
| 3i | 16 (7.0) | 18 (9.0) | 14 (3.0) | 14 (4.0) | 25 (4.0) | 23 (10) |
| 3j | 16 (9.0) | 18 (7.5) | 15 (2.5) | 14 (6.0) | 25 (4.0) | 24 (10) |
| 3k | 14 (8.0) | 16 (12) | 11 (3.0) | 10 (10) | 21 (8.0) | 20 (14) |
| Levofloxacin | 13 (0.4) | 15 (5.0) | 10 (1.25) | — | — | — |
| Clotrimazole | — | — | — | 20 (0.5) | 24 (0.4) | 36 (1.25) |
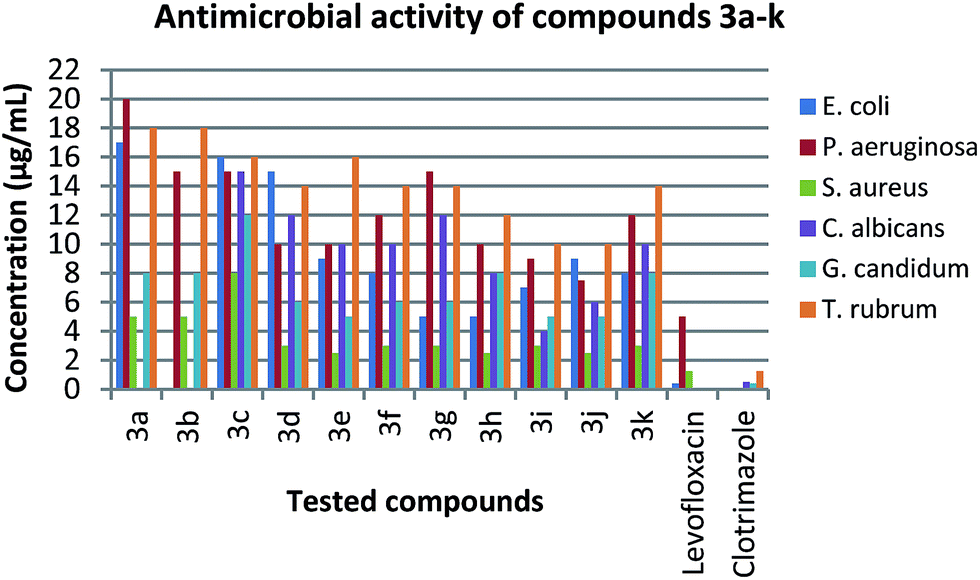 | ||
| Fig. 4 Comparison of MIC (μg mL−1) of the synthesized compounds 3a–k with standard microorganisms. | ||
In the case of antibacterial activity (Table 4), the experimental results indicated a remarkable activity of the synthesized compounds. Compounds 3g and 3h exhibited the highest activity MIC 5.0 μg mL−1 against Escherichia coli (−ve), while compounds 3e, 3f, 3i, 3j and 3k showed moderate to strong activity. In addition, 3a, 3c, 3d showed somewhat inferior activity, compared to levofloxacin (MIC 0.4 μg mL−1). In the case of Pseudomonas aeruginosa (−ve), compounds 3d, 3e, 3f, 3g, 3h, 3i, 3j and 3k were found to be the most active derivatives. However compounds 3a, 3b and 3c showed moderate activity, compared to levofloxacin (MIC 5.0 μg mL−1). Furthermore, compounds 3d, 3e, 3f, 3g, 3h, 3i, 3j and 3k showed strong activity with MIC 3.0, 2.5, 3.0, 3.0, 2.5, 3.0, 2.5 and 3.0 μg mL−1 respectively against Staphylococcus aureus (+ve). While, compounds 3a, 3b and 3c were showed moderate activity compared to the antibacterial reference of levofloxacin (MIC 1.25 μg mL−1).
In the same manner, all the synthesized compounds showed a noteworthy activity against different strains of fungi (Table 4). Compounds 3h, 3i and 3j showed the highest activity with MIC 6.0, 4.0 and 6.0 μg mL−1 against Candida albicans, respectively. However, compounds 3c, 3d, 3e, 3f, 3g and 3k showed good to moderate activity, while compounds 3a and 3b did not show antifungal activity as compared with clotrimazole (MIC 0.5 μg mL−1). In the case of Geotrichum candidum, compounds 3d, 3e, 3f, 3g, 3i and 3j showed strong activity with MIC 5.0, 4.0, 5.0, 5.0, 4.0 and 4.0 μg mL−1 respectively, while compounds 3a, 3b, 3c, 3h and 3k were showed moderate activity, compared to clotrimazole (MIC 0.4 μg mL−1). Additionally, compounds 3d, 3f, 3g, 3h, 3i and 3j showed excellent activity with MIC 12, 12, 12, 12, 10 and 10 against Trichophyton rubrum, respectively, while compounds 3a, 3b, 3c, 3e and 3k showed moderate activity as compared to the reference agent clotrimazole (MIC 1.25 μg mL−1).
5. Structure-activity relationship (SAR)
The structure-activity studies showed that the nature of primary amine moiety has a profound effect on the microbial inhibitory activity (Table 4) (Fig. 4). The data in Table 4 indicated a strong activity against Gram-positive bacteria. The tested compounds showed same order of Staphylococcus aureus inhibition compared to the reference drug levofloxacin (MIC of 2.5–8.0 μg mL−1 vs. 1.25 μg mL−1, respectively). The introduction of aromatic moieties such as benzyl, phenyl and naphthyl (3d, 3e and 3k) enhanced the inhibition activity generally 2–3 times more than the aliphatic containing analogues (3a–c). It can be also noted that the introduction of substituent alkoxy, alkyl or halogen groups onto phenyl ring (3f–j) returned similar levels of Gram-positive inhibition. The data also showed the ability of the tested compounds to inhibit the Gram-negative Pseudomonas aeruginosa at MIC levels comparable to the reference levofloxacin. Similar to what was noted for Gram-positive bacteria, aromatic derivatives are generally more active against Pseudomonas aeruginosa with well-tolerated of substituent variations. However, the tested derivatives were 1–2 orders less active against Escherichia coli compared to levofloxacin.Similarly, the newly synthesized compounds showed varying degrees of fungal inhibitory activity (Table 4) (Fig. 4). In general, the inhibition activity against Candida albicans, Geotrichum candidum and Trichophyton rubrum indicated that the introduction of aromatic amine (3d–k) improved the activity 1–3 times more than aliphatic amines (3a–c). Further, SAR studies revealed that aromatic amines containing halogen atom (3i and 3j) showed the highest inhibitory activity against all fungi, which was approximately 1 order less that of the reference drug clotrimazole. Introduction of phenyl, benzyl and naphthyl moiety (3d, 3e and 3k) inhibited the Geotrichum candidum and Trichophyton rubrum at MIC levels comparable to halogen containing analogues, whereas 2 times less against Candida albicans. It was also noted that replacement of halogen atom by alkoxy or alkyl group (3f and 3h) slightly reduced the activity against all types of fungi. Changing the substituent position showed that methyl group in meta-position (3g) reduced the activity 2 times than para-position (3h) against Candida albicans, while increased the activity 2 times against Geotrichum candidum and remaining constant against Trichophyton rubrum.
6. Conclusions
In summary, we have developed an efficient, rapid and regioselective synthesis of 2,5,7,9,11-pentaazaphenalenes via a quadruple Mannich reaction of readily available 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one, formaldehyde and primary amines in ethanol at room temperature for 1 h. The reaction products were easily isolated in very good to excellent yield by filtration and then purified by recrystallization from the proper solvent. The electronic energy calculations of the expected isomeric products indicated that the most stable and favourable isomeric products is 2,5,7,9,11-pentaazaphenalenes rather than 3,5,8,10,11-pentaazaanthracenes or 2,5,7,8,10-pentaza-phenanthrenes. In addition, all the synthesized products showed high antimicrobial activity against Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Candida albicans, Geotrichum candidum and Trichophyton rubrum. Therefore, this elegant strategy can be utilized in the synthesis of a library of compounds medicinally and pharmaceutically significant. The formation of single crystals of the synthesized compounds for X-ray analysis is in progress.7. Experimental
7.1. Chemistry
All reagents and solvents were used as received from commercial sources without further purification. Melting points were determined using a Gallencamp melting point apparatus. Infrared spectra were recorded using KBr discs on a Perkin-Elmer 1430 FT-IR spectrometer at room temperature. 1H-NMR and 13C-NMR spectra were recorded at room temperature using DMSO-d6 as a solvent on a Bruker Advance 400 NMR spectrometer (Centre of Analysis and Synthesis, Department of Chemistry, Lund University, Sweden) or a Varian EM-390 90 MHz spectrometer (Spectral Unit, Chemistry department, Assiut University, Egypt). Chemical shifts were donated in ppm on δ scale, relative to TMS as internal standard and coupling constants (J) are given in Hz. Mass spectra were recorded on a JEOL JMS-600 mass spectrometer using a direct inlet system. Elemental analyses were measured on a Perkin-Elmer 240 C elemental analyzer.General procedure. 6-Amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (1) (4 mmol, 0.572 g) was added at room temperature to a stirred solution of primary amines (2a–k) (8 mmol) and excess of formaldehyde solution (35 wt% in H2O, 1 mL) in ethanol (20 mL). The reaction mixture was continuously stirred at room temperature for 1 h, and then the resulting precipitate was collected by filtration and washed several times with ethanol. The crude product was purified by recrystallization from the proper solvent to afford the desired product (3a–k) in 82–95% yields. All products dissolved in alcoholic KOH (5%) and reprecipitated again by addition of diluted acetic acid.
O), 1590 (CN), 1510 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 0.87 (t, 6H, J = 2 Hz, 2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3CH2), 2.48 (q, 4H, J = 2 Hz, 2C2CH3), 3.46 (s, 2H, CCH2N), 4.06 (s, 2H, NCH2N), 4.24 (s, 2H, NCH2N), 5.21 (s, 2H, NCH2N), 11.88 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 175.02 (CS), 162.08 (CO), 154.78 (CC), 83.32 (CC), 67.69 (NCH2N), 66.96 (NCH2N), 64.75 (NCH2N), 58.25 (CCH2N), 46.89 (N
3CH2), 2.48 (q, 4H, J = 2 Hz, 2C2CH3), 3.46 (s, 2H, CCH2N), 4.06 (s, 2H, NCH2N), 4.24 (s, 2H, NCH2N), 5.21 (s, 2H, NCH2N), 11.88 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 175.02 (CS), 162.08 (CO), 154.78 (CC), 83.32 (CC), 67.69 (NCH2N), 66.96 (NCH2N), 64.75 (NCH2N), 58.25 (CCH2N), 46.89 (N![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CH3), 46.47 (NH2CH3), 20.69 (NCH2H3), 21.05 (NCH2H3). MS (EI, 70 eV): m/z (%) = 280.28, 211.02, 180.95, 126.01, 29.02. Anal. calcd. for C12H19N5OS (281.38): C, 51.22; H, 6.81; N, 24.89; S, 11.40%; found: C, 51.30; H, 6.78; N, 24.95; S, 11.36%.O), 1593 (CN), 1517 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 0.88 (d, 12H, J = 2 Hz, 2(C3)2CH), 2.20 (m, 2H, J = 2 Hz, 2(CH3)2C), 3.05 (d, 4H, J = 2 Hz, 2C2CH), 3.47 (s, 2H, CCH2N), 4.09 (s, 2H, NCH2N), 4.27 (s, 2H, NCH2N), 5.23 (s, 2H, NCH2N), 11.91 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.75 (CS), 158.62 (CO), 149.05 (CC), 78.62 (CC), 68.09 (NCH2N), 67.07 (NCH2N), 64.84 (NCH2N), 60.42 (CH3)2CHH2, 60.14 (CH3)2CHH2, 46.98 (CCH2N), 26.43 (CH3)2H, 26.40 (CH3)2H, 20.92 (H3)2CH, 20.45 (H3)2CH. MS (EI, 70 eV): m/z (%) = 336.58, 238.01, 179.91, 125.88, 57.99. Anal. calcd. for C16H27N5OS (337.48): C, 56.94; H, 8.06; N, 20.75; S, 9.50%; found: C, 56.88; H, 8.12; N, 20.82; S, 9.41%.O), 1593 (CN), 1516 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 0.88 (t, 6H, J = 2 Hz, 2C3CH2), 1.28 (m, 8H, 2CH3C2C2), 2.51 (t, 4H, J = 2 Hz, 2NC2), 3.45 (s, 2H, CCH2N), 4.05 (s, 2H, NCH2N), 4.25 (s, 2H, NCH2N), 5.20 (s, 2H, NCH2N), 11.85 (s, 1H, NH). Anal. calcd. for C16H27N5OS (337.48): C, 56.94; H, 8.06; N, 20.75; S, 9.50%; found: C, 56.99; H, 8.10; N, 20.66; S, 9.42%.O), 1588 (CN), 1516 (CC) cm−1. 1H-NMR (90 MHz, DMSO-d6): δ = 3.46 (s, 2H, CCH2N), 3.71 (s, 2H, NCH2-benzyl), 3.89 (s, 2H, NCH2-benzyl), 4.03 (s, 2H, NCH2N), 4.16 (s, 2H, NCH2N), 5.15 (s, 2H, NCH2N), 7.35 (br., s, 10H, H-arom.), 11.78 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.25 (CS), 158.21 (CO), 147.59 (CC), 138.18 (C-arom.), 137.02 (C-arom.), 128.99 (2CH-arom.), 128.73 (2CH-arom.), 128.42 (2CH-arom.), 128.31 (2CH-arom.), 127.59 (CH-arom.), 127.19 (CH-arom.), 82.96 (CC), 66.22 (NCH2N), 66.01 (NCH2N), 63.61 (NCH2N), 55.83 (NH2benzyl), 54.40 (NH2benzyl), 45.41 (CCH2N). MS (EI, 70 eV): m/z (%) = 331.38, 285.47, 179.70, 134.80, 83.85. Anal. calcd. for C22H23N5OS (405.52): C, 65.16; H, 5.72; N, 17.27; S, 7.91%; found: C, 65.08; H, 5.76; N, 17.34; S, 7.84%.O), 1589 (CN), 1489 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 4.10 (s, 2H, CCH2N), 4.85 (s, 2H, NCH2N), 5.05 (s, 2H, NCH2N), 5.87 (s, 2H, NCH2N), 6.86–7.27 (m, 10H, H-arom.), 12.28 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.43 (CS), 158.05 (CO), 149.32 (C-arom.), 148.60 (C-arom.), 146.12 (CC), 129.91 (2CH-arom.), 129.50 (2CH-arom.), 122.67 (CH-arom.), 121.07 (CH-arom.), 118.08 (2CH-arom.), 117.78 (2CH-arom.), 85.27 (CC), 64.46 (NCH2N), 63.60 (NCH2N), 63.15 (NCH2N), 45.16 (CCH2N). Anal. calcd. for C20H19N5OS (377.46): C, 63.64; H, 5.07; N, 18.55; S, 8.49%; found: C, 63.71; H, 4.98; N, 18.62; S, 8.41%.O), 1597 (CN), 1508 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 3.69 (s, 6H, 2OCH3), 4.05 (s, 2H, CCH2N), 4.74 (s, 2H, NCH2N), 4.91 (s, 2H, NCH2N), 5.76 (s, 2H, NCH2N), 6.71–6.79 (m, 8H, H-arom.), 11.97 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.49 (CS), 158.08 (CO), 155.27 (C-arom.), 154.34 (C-arom.), 148.53 (CC), 142.22 (C-arom.), 139.72 (C-arom.), 119.94 (2CH-arom.), 119.73 (2CH-arom.), 115.06 (2CH-arom.), 114.76 (2CH-arom.), 84.94 (CC), 65.46 (NCH2N), 65.08 (NCH2N), 64.08 (NCH2N), 55.58 (OCH3), 45.72 (CCH2N). MS (EI, 70 eV): m/z (%) = 437.12, 406.07, 334.05, 289.12, 198.05, 135.06, 120.04, 108.12, 92.04. Anal. calcd. for C22H23N5O3S (437.51): C, 60.39; H, 5.30; N, 16.01; S, 7.33%; found: C, 60.42; H, 5.38; N, 15.96; S, 7.28%.O), 1602 (CN), 1578 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 2.20 (s, 6H, 2CH3), 4.07 (s, 2H, CCH2N), 4.78 (s, 2H, NCH2N), 5.03 (s, 2H, NCH2N), 5.83 (s, 2H, NCH2N), 6.62–7.16 (m, 8H, H-arom.), 12.08 (s, 1H, NH). Anal. calcd. for C22H23N5OS (405.52): C, 65.16; H, 5.72; N, 17.27; S, 7.91%; found: C, 65.22; H, 5.68; N, 17.33; S, 7.85%.O), 1636 (CN), 1589 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 2.18 (s, 6H, 2CH3), 4.00 (s, 2H, CCH2N), 4.70 (s, 2H, NCH2N), 4.85 (s, 2H, NCH2N), 5.70 (s, 2H, NCH2N), 6.65–6.95 (m, 8H, H-arom.), 11.95 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.42 (CS), 158.07 (CO), 149.30 (C-arom.), 148.51 (C-arom.), 144.14 (CC), 136.60 (C-arom.), 134.46 (C-arom.), 127.90 (2CH-arom.), 127.71 (2CH-arom.), 118.90 (2CH-arom.), 117.71 (2CH-arom.), 84.21 (CC), 64.41 (NCH2N), 63.58 (NCH2N), 63.12 (NCH2N), 45.13 (CCH2N), 20.92 (2CH3). MS (EI, 70 eV): m/z (%) = 404.93, 390.11, 271.18, 182.08, 119.04, 102.52. Anal. calcd. for C22H23N5OS (405.52): C, 65.16; H, 5.72; N, 17.27; S, 7.91%; found: C, 65.09; H, 5.64; N, 17.36; S, 7.96%.O), 1601 (CN), 1548 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 4.08 (s, 2H, CCH2N), 4.82 (s, 2H, NCH2N), 5.10 (s, 2H, NCH2N), 5.80 (s, 2H, NCH2N), 6.86 (d, 4H, J = 4 Hz, H-arom.), 7.32 (d, 4H, J = 4 Hz, H-arom.), 12.05 (s, 1H, NH). MS (EI, 70 eV): m/z (%) = 445.00, 292.00, 290.99, 141.04, 140.03, 139.03, 111.00, 92.05, 75.03. Anal. calcd. for C20H17Cl2N5OS (445.05): C, 53.82; H, 3.84; N, 15.69; S, 7.18%; found: C, 53.89; H, 3.78; N, 15.74; S, 7.14%.O), 1600 (CN), 1510 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 4.11 (s, 2H, CCH2N), 4.84 (s, 2H, NCH2N), 5.02 (s, 2H, NCH2N), 5.82 (s, 2H, NCH2N), 6.85 (d, 4H, J = 4 Hz, H-arom.), 7.36 (d, 4H, J = 4 Hz, H-arom.), 12.07 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.57 (CS), 158.07 (CO), 148.45 (C-arom.), 147.73 (C-arom.), 145.45 (CC), 132.53 (2CH-arom.), 132.09 (2CH-arom.), 120.17 (2CH-arom.), 119.96 (2CH-arom.), 114.62 (C-arom.), 112.75 (C-arom.), 85.30 (CC), 64.33 (NCH2N), 63.85 (NCH2N), 63.37 (NCH2N), 44.99 (CCH2N). MS (EI, 70 eV): m/z (%) = 534.16, 350.93, 339.06, 337.06, 289.96, 183.97, 168.01, 154.96, 156.00, 108.99, 92.99, 78.99. Anal. calcd. for C20H17Br2N5OS (534.95): C, 44.88; H, 3.20; N, 13.08; S, 5.99%; found: C, 44.94; H, 3.22; N, 12.97; S, 6.20%.O), 1590 (CN), 1514 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 3.72 (d, 2H, J = 12 Hz, CCH2N), 4.70 (t, J = 12 Hz, 2H, NCH2N), 4.92 (t, 2H, J = 12 Hz, NCH2N), 6.35 (s, 2H, NCH2N), 7.21–7.89 (m, 14H, H-arom.), 12.16 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 179.59 (CS), 169.53 (CO), 144.39 (C-arom.), 141.35 (C-arom.), 135.67 (CC), 133.25 (C-arom.), 133.10 (C-arom.), 130.96 (CH-arom.), 128.75 (2CH-arom.), 128.39 (CH-arom.), 127.74 (C-arom.), 127.28 (C-arom.), 126.65 (2CH-arom.), 125.50 (CH-arom.), 125.00 (CH-arom.), 122.92 (CH-arom.), 121.80 (CH-arom.), 121.15 (CH-arom.), 118.63 (CH-arom.), 111.84 (CH-arom.), 109.89 (CH-arom.), 70.81 (CC), 49.94 (NCH2N), 48.82 (NCH2N), 47.19 (NCH2N), 35.96 (CCH2N). Anal. calcd. for C20H17Cl2N5OS (445.05): C, 53.82; H, 3.84; N, 15.69; S, 7.18%; found: C, 53.89; H, 3.78; N, 15.74; S, 7.14%.
H2CH3), 46.47 (NH2CH3), 20.69 (NCH2H3), 21.05 (NCH2H3). MS (EI, 70 eV): m/z (%) = 280.28, 211.02, 180.95, 126.01, 29.02. Anal. calcd. for C12H19N5OS (281.38): C, 51.22; H, 6.81; N, 24.89; S, 11.40%; found: C, 51.30; H, 6.78; N, 24.95; S, 11.36%.O), 1593 (CN), 1517 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 0.88 (d, 12H, J = 2 Hz, 2(C3)2CH), 2.20 (m, 2H, J = 2 Hz, 2(CH3)2C), 3.05 (d, 4H, J = 2 Hz, 2C2CH), 3.47 (s, 2H, CCH2N), 4.09 (s, 2H, NCH2N), 4.27 (s, 2H, NCH2N), 5.23 (s, 2H, NCH2N), 11.91 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.75 (CS), 158.62 (CO), 149.05 (CC), 78.62 (CC), 68.09 (NCH2N), 67.07 (NCH2N), 64.84 (NCH2N), 60.42 (CH3)2CHH2, 60.14 (CH3)2CHH2, 46.98 (CCH2N), 26.43 (CH3)2H, 26.40 (CH3)2H, 20.92 (H3)2CH, 20.45 (H3)2CH. MS (EI, 70 eV): m/z (%) = 336.58, 238.01, 179.91, 125.88, 57.99. Anal. calcd. for C16H27N5OS (337.48): C, 56.94; H, 8.06; N, 20.75; S, 9.50%; found: C, 56.88; H, 8.12; N, 20.82; S, 9.41%.O), 1593 (CN), 1516 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 0.88 (t, 6H, J = 2 Hz, 2C3CH2), 1.28 (m, 8H, 2CH3C2C2), 2.51 (t, 4H, J = 2 Hz, 2NC2), 3.45 (s, 2H, CCH2N), 4.05 (s, 2H, NCH2N), 4.25 (s, 2H, NCH2N), 5.20 (s, 2H, NCH2N), 11.85 (s, 1H, NH). Anal. calcd. for C16H27N5OS (337.48): C, 56.94; H, 8.06; N, 20.75; S, 9.50%; found: C, 56.99; H, 8.10; N, 20.66; S, 9.42%.O), 1588 (CN), 1516 (CC) cm−1. 1H-NMR (90 MHz, DMSO-d6): δ = 3.46 (s, 2H, CCH2N), 3.71 (s, 2H, NCH2-benzyl), 3.89 (s, 2H, NCH2-benzyl), 4.03 (s, 2H, NCH2N), 4.16 (s, 2H, NCH2N), 5.15 (s, 2H, NCH2N), 7.35 (br., s, 10H, H-arom.), 11.78 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.25 (CS), 158.21 (CO), 147.59 (CC), 138.18 (C-arom.), 137.02 (C-arom.), 128.99 (2CH-arom.), 128.73 (2CH-arom.), 128.42 (2CH-arom.), 128.31 (2CH-arom.), 127.59 (CH-arom.), 127.19 (CH-arom.), 82.96 (CC), 66.22 (NCH2N), 66.01 (NCH2N), 63.61 (NCH2N), 55.83 (NH2benzyl), 54.40 (NH2benzyl), 45.41 (CCH2N). MS (EI, 70 eV): m/z (%) = 331.38, 285.47, 179.70, 134.80, 83.85. Anal. calcd. for C22H23N5OS (405.52): C, 65.16; H, 5.72; N, 17.27; S, 7.91%; found: C, 65.08; H, 5.76; N, 17.34; S, 7.84%.O), 1589 (CN), 1489 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 4.10 (s, 2H, CCH2N), 4.85 (s, 2H, NCH2N), 5.05 (s, 2H, NCH2N), 5.87 (s, 2H, NCH2N), 6.86–7.27 (m, 10H, H-arom.), 12.28 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.43 (CS), 158.05 (CO), 149.32 (C-arom.), 148.60 (C-arom.), 146.12 (CC), 129.91 (2CH-arom.), 129.50 (2CH-arom.), 122.67 (CH-arom.), 121.07 (CH-arom.), 118.08 (2CH-arom.), 117.78 (2CH-arom.), 85.27 (CC), 64.46 (NCH2N), 63.60 (NCH2N), 63.15 (NCH2N), 45.16 (CCH2N). Anal. calcd. for C20H19N5OS (377.46): C, 63.64; H, 5.07; N, 18.55; S, 8.49%; found: C, 63.71; H, 4.98; N, 18.62; S, 8.41%.O), 1597 (CN), 1508 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 3.69 (s, 6H, 2OCH3), 4.05 (s, 2H, CCH2N), 4.74 (s, 2H, NCH2N), 4.91 (s, 2H, NCH2N), 5.76 (s, 2H, NCH2N), 6.71–6.79 (m, 8H, H-arom.), 11.97 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.49 (CS), 158.08 (CO), 155.27 (C-arom.), 154.34 (C-arom.), 148.53 (CC), 142.22 (C-arom.), 139.72 (C-arom.), 119.94 (2CH-arom.), 119.73 (2CH-arom.), 115.06 (2CH-arom.), 114.76 (2CH-arom.), 84.94 (CC), 65.46 (NCH2N), 65.08 (NCH2N), 64.08 (NCH2N), 55.58 (OCH3), 45.72 (CCH2N). MS (EI, 70 eV): m/z (%) = 437.12, 406.07, 334.05, 289.12, 198.05, 135.06, 120.04, 108.12, 92.04. Anal. calcd. for C22H23N5O3S (437.51): C, 60.39; H, 5.30; N, 16.01; S, 7.33%; found: C, 60.42; H, 5.38; N, 15.96; S, 7.28%.O), 1602 (CN), 1578 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 2.20 (s, 6H, 2CH3), 4.07 (s, 2H, CCH2N), 4.78 (s, 2H, NCH2N), 5.03 (s, 2H, NCH2N), 5.83 (s, 2H, NCH2N), 6.62–7.16 (m, 8H, H-arom.), 12.08 (s, 1H, NH). Anal. calcd. for C22H23N5OS (405.52): C, 65.16; H, 5.72; N, 17.27; S, 7.91%; found: C, 65.22; H, 5.68; N, 17.33; S, 7.85%.O), 1636 (CN), 1589 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 2.18 (s, 6H, 2CH3), 4.00 (s, 2H, CCH2N), 4.70 (s, 2H, NCH2N), 4.85 (s, 2H, NCH2N), 5.70 (s, 2H, NCH2N), 6.65–6.95 (m, 8H, H-arom.), 11.95 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.42 (CS), 158.07 (CO), 149.30 (C-arom.), 148.51 (C-arom.), 144.14 (CC), 136.60 (C-arom.), 134.46 (C-arom.), 127.90 (2CH-arom.), 127.71 (2CH-arom.), 118.90 (2CH-arom.), 117.71 (2CH-arom.), 84.21 (CC), 64.41 (NCH2N), 63.58 (NCH2N), 63.12 (NCH2N), 45.13 (CCH2N), 20.92 (2CH3). MS (EI, 70 eV): m/z (%) = 404.93, 390.11, 271.18, 182.08, 119.04, 102.52. Anal. calcd. for C22H23N5OS (405.52): C, 65.16; H, 5.72; N, 17.27; S, 7.91%; found: C, 65.09; H, 5.64; N, 17.36; S, 7.96%.O), 1601 (CN), 1548 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 4.08 (s, 2H, CCH2N), 4.82 (s, 2H, NCH2N), 5.10 (s, 2H, NCH2N), 5.80 (s, 2H, NCH2N), 6.86 (d, 4H, J = 4 Hz, H-arom.), 7.32 (d, 4H, J = 4 Hz, H-arom.), 12.05 (s, 1H, NH). MS (EI, 70 eV): m/z (%) = 445.00, 292.00, 290.99, 141.04, 140.03, 139.03, 111.00, 92.05, 75.03. Anal. calcd. for C20H17Cl2N5OS (445.05): C, 53.82; H, 3.84; N, 15.69; S, 7.18%; found: C, 53.89; H, 3.78; N, 15.74; S, 7.14%.O), 1600 (CN), 1510 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 4.11 (s, 2H, CCH2N), 4.84 (s, 2H, NCH2N), 5.02 (s, 2H, NCH2N), 5.82 (s, 2H, NCH2N), 6.85 (d, 4H, J = 4 Hz, H-arom.), 7.36 (d, 4H, J = 4 Hz, H-arom.), 12.07 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 173.57 (CS), 158.07 (CO), 148.45 (C-arom.), 147.73 (C-arom.), 145.45 (CC), 132.53 (2CH-arom.), 132.09 (2CH-arom.), 120.17 (2CH-arom.), 119.96 (2CH-arom.), 114.62 (C-arom.), 112.75 (C-arom.), 85.30 (CC), 64.33 (NCH2N), 63.85 (NCH2N), 63.37 (NCH2N), 44.99 (CCH2N). MS (EI, 70 eV): m/z (%) = 534.16, 350.93, 339.06, 337.06, 289.96, 183.97, 168.01, 154.96, 156.00, 108.99, 92.99, 78.99. Anal. calcd. for C20H17Br2N5OS (534.95): C, 44.88; H, 3.20; N, 13.08; S, 5.99%; found: C, 44.94; H, 3.22; N, 12.97; S, 6.20%.O), 1590 (CN), 1514 (CC) cm−1. 1H-NMR (400 MHz, DMSO-d6): δ = 3.72 (d, 2H, J = 12 Hz, CCH2N), 4.70 (t, J = 12 Hz, 2H, NCH2N), 4.92 (t, 2H, J = 12 Hz, NCH2N), 6.35 (s, 2H, NCH2N), 7.21–7.89 (m, 14H, H-arom.), 12.16 (s, 1H, NH). 13C-NMR (400 MHz, DMSO-d6): δ = 179.59 (CS), 169.53 (CO), 144.39 (C-arom.), 141.35 (C-arom.), 135.67 (CC), 133.25 (C-arom.), 133.10 (C-arom.), 130.96 (CH-arom.), 128.75 (2CH-arom.), 128.39 (CH-arom.), 127.74 (C-arom.), 127.28 (C-arom.), 126.65 (2CH-arom.), 125.50 (CH-arom.), 125.00 (CH-arom.), 122.92 (CH-arom.), 121.80 (CH-arom.), 121.15 (CH-arom.), 118.63 (CH-arom.), 111.84 (CH-arom.), 109.89 (CH-arom.), 70.81 (CC), 49.94 (NCH2N), 48.82 (NCH2N), 47.19 (NCH2N), 35.96 (CCH2N). Anal. calcd. for C20H17Cl2N5OS (445.05): C, 53.82; H, 3.84; N, 15.69; S, 7.18%; found: C, 53.89; H, 3.78; N, 15.74; S, 7.14%.7.2. Electronic energy and atomic charge calculations
All electronic energy calculations of intermediate tautomers (6a–k, 7a–k and 8a–k) and of isomeric products (3a–k, 4a–k and 5a–k) are performed using the B3LYP functional of the Gaussian 09 software package with 6-31+G(d,p) basis set.32 The atomic charge calculations were performed using the natural population analysis method using B3LYP/6-31+G(d,p).7.3. Antimicrobial activity
The three bacterial strains and the three fungi strains used in the present study were isolated from some cases of human dermatophytosis (Assiut university Mycological center). The target strains were allowed to grow in sterilized 9 cm Petri-dishes containing Sabourand's dextrose agar (SDA) supplementary with 0.05% of chloramphenicol.33 From these cultures, 10 mm diameter agar discs containing spores were transferred to screw-topped vials, followed by addition of 20 mL sterilized water. After shaking, 1 mL of the spore suspension was seeded into sterilized Petri-dishes followed by addition of 15 mL of liquefied SDA medium and then let to solidify.The synthesized compounds 3a–k to be screened, were dissolved in DMSO to give a solution of 2% concentration. Filter paper discs (Whatman No. 3) with approximately 5 mm in diameter were soaked with 15 mL of the tested compound solutions and then placed on the surface of the previously prepared agar plates which seeded by the tested bacteria and fungi. Each disc was immersed down to ensure complete contact with the agar surface. Then the agar plates were incubated at 37 °C for 16–18 h for bacteria and at room temperature for 4 days for fungi. The diameters of the zones of compound inhibition were measured and listed in Table 4. A similar procedure was performed for commercial antibiotics levofloxacin and clotrimazole which were used as positive control for bacteria and fungi, respectively.
The minimum inhibitory concentration (MIC) of each compound was determined by micro dilution method.34 The biologically active compounds were serially diluted in DMSO and incubated with 10 mL broth tubes inoculated with the test culture for 24 h. MIC of each compound was taken as the lowest concentration (μg mL−1) that did not give any visible bacteria or fungi growth (Table 4).
References
- (a) A. Kaur, M. Kaur and B. Singh, J. Heterocycl. Chem., 2014, 52, 827–833 CrossRef; (b) D. J. Ramon and M. Yus, Angew. Chem., 2005, 117, 1628–1661 (Angew. Chem., Int. Ed., 2005, 44, 1602–1634) CrossRef.
- (a) E. Ruijter, R. Scheffelaar and R. V. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed; (b) T. J. Muller, Beilstein J. Org. Chem., 2011, 7, 960–961 CrossRef PubMed; (c) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114(16), 8323–8359 CrossRef CAS PubMed.
- (a) A. Dçmling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (b) D. Tejedor and F. G. Tellado, Chem. Soc. Rev., 2007, 36, 484–491 RSC; (c) C. Simon, T. Con-stantieux and J. Rodriguez, Eur. J. Org. Chem., 2004, 4957–4980 CrossRef CAS.
- (a) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC; (b) C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51–80 CrossRef CAS PubMed; (c) F. Lieby-Muller, T. Constantieux and J. Rodriguez, J. Am. Chem. Soc., 2005, 127, 17176–17177 CrossRef CAS PubMed.
- (a) R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123–131 CrossRef CAS; (b) G. H. Posner, Chem. Rev., 1986, 86, 831–844 CrossRef CAS; (c) L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed. Engl., 1993, 32, 131–163 CrossRef; (d) R. A. Bunce, Tetrahedron, 1995, 51, 13103–13159 CrossRef CAS.
- (a) B. S. Holla, M. Mahalinga, M. S. Karthikeyan, P. M. Akberali and N. S. Shetty, Bioorg. Med. Chem., 2006, 14, 2040–2047 CrossRef CAS PubMed; (b) N. Ingarsal, G. Saravanan, P. Amutha and S. Nagarajan, Eur. J. Med. Chem., 2007, 42, 517–520 CrossRef CAS PubMed.
- E. Wyrzykiewicz, G. Bartkowiak and B. Kedzia, Farmaco, 1993, 48, 979–988 CAS.
- (a) S. M. Sondhi, N. Singh, M. Johar and A. Kumar, Bioorg. Med. Chem., 2005, 13, 6158–6166 CrossRef CAS PubMed; (b) K. M. Amin, M. M. Hanna, H. E. Abo-Youssef and R. F. George, Eur. J. Med. Chem., 2009, 1–13 Search PubMed.
- L. Cordeu, E. Cubedo, E. Bandrés, A. Rebollo, X. Sáenz, H. Chozas, M. V. Domínguez, M. Echeverría, B. Mendivil, C. Sanmartin, J. A. Palop, M. Font and J. García-Foncillas, Bioorg. Med. Chem., 2007, 15, 1659–1669 CrossRef CAS PubMed.
- D. L. Powers, J. W. Sowell Sr, J. J. Freeman and J. W. Kosh, J. Pharm. Sci., 2006, 69, 473–475 CrossRef.
- T. Kimura, S. Teraoka, J. Kuze, K. Watanabe, S. Kondo, I. Ho and I. Yamamoto, Nucleic Acids Symp. Ser., 1993, 29, 51–52 CAS.
- (a) M. Pemmsin, C. Lnu-Due, F. Hoguet, C. Gaultier and J. Narcisse, Eur. J. Chem., 1988, 23, 534–541 Search PubMed; (b) A. B. A. El-Gazzar, M. M. El-Enany and M. N. Mahmoud, Bioorg. Med. Chem., 2008, 16, 3261–3273 CrossRef CAS PubMed.
- J. L. Bernier, J. P. Henichart, V. Warin and F. Baert, J. Pharm. Sci., 2006, 69, 1343–1345 CrossRef.
- P. A. S. Smith and R. O. Kan, J. Org. Chem., 1964, 29, 2261–2265 CrossRef CAS.
- P. G. Hoggard, S. D. Sales, S. Kewn, D. Sunderland, S. H. Khoo, C. A. Hart and D. J. Back, Antiviral Chem. Chemother., 2000, 11, 353–358 CrossRef CAS.
- R. van Leeuwen, C. Katlama, V. Kitchen, C. A. Boucher, R. Tubiana, M. McBride, D. Ingrand, J. Weber, A. Hill, H. McDade and S. A. Danner, J. Infect. Dis., 1995, 171(5), 1166–1171 CrossRef CAS PubMed.
- E. De Clercq, Anticancer Res., 1986, 6(4), 549–556 CAS.
- I. Homminga, C. M. Zwaan, C. Y. Manz, C. Parker, S. Bantia, W. K. Smits, F. Higginbotham, R. Pieters and J. P. P. Meijerink, Blood, 2011, 118(8), 2184–2190 CrossRef CAS PubMed.
- A. Gangjee, O. Aldair and S. F. Queener, J. Med. Chem., 1999, 42, 2447–2455 CrossRef CAS PubMed.
- A. Gang Jee, J. Yu, L. R. Kisliuk, H. W. Halie, G. Sobrero and J. J. McGuire, J. Med. Chem., 2003, 46, 591 CrossRef CAS PubMed.
- S. Furuya and T. Ohtaki, Eur. Pat. Appl. Ep., 1994, 608565 (Chem. Abstr., 1994, 121, 205395w) Search PubMed.
- J. I. DeGraw, P. H. Christie, W. T. Clowell and F. M. Sirotnak, J. Med. Chem., 1992, 35, 320–324 CrossRef CAS PubMed.
- L. B. Gordon, E. A. Donald, S. L. Banitt, L. B. Kenneth, A. M. Stephen, J. R. Palmer and J. M. Tustin, J. Med. Chem., 1995, 38, 4161–4163 CrossRef.
- M. Arend, B. Westermann and N. Risch, Angew. Chem., 1998, 110, 1096–1122 CrossRef.
- Z. Wang, T. You and H. Shi, Molecules, 1996, 1, 89–92 CrossRef CAS.
- H. A. H. El-Sherief, Z. A. Hozien, A. F. M. EL-Mahdy and A. A. O. Sarhan, J. Heterocycl. Chem., 2010, 47(6), 1294–1302 CrossRef CAS.
- (a) Z. A. Hozien, A. A. O. Sarhan, H. A. H. El-Sherief and A. M. Mahmoud, Z. Naturforsch., B: J. Chem. Sci., 1997, 52, 1401–1412 CAS; (b) A. M. Mahmoud, H. A. H. El-Sherief, O. M. A. Habib and A. A. O. Sarhan, Phosphorus, Sulfur Silicon Relat. Elem., 2007, 182, 1757–1776 CrossRef CAS.
- A. A. O. Sarhan, S. H. Abdel-Hafez, H. A. H. El -Sherief and T. Aboel-Fadl, Synth. Commun., 2006, 36, 987–996 CrossRef CAS.
- M. S. Mohamed, S. M. Awad and A. I. Sayed, Molecules, 2010, 15, 1882–1890 CrossRef CAS PubMed.
- J. J. Marugan, W. Zheng, O. Motabar, N. Southall, E. Goldin, E. Sidransky, R. A. Aungst, K. Liu, S. K. Sadhukhan and C. P. Austin, Eur. J. Med. Chem., 2010, 45(5), 1880–1897 CrossRef CAS PubMed.
- T. j. Delia, J. P. Scovill, W. D. Munslow and J. H. Burchalter, J. Med. Chem., 1976, 19(2), 344–346 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Al-Doory, Laboratory Medical Mycology, Lea and Febiger, Philadelphia, 1980, vol. 20, suppl. 19, pp. 219–241 Search PubMed.
- Practical Medicinal Microbiology, ed. J. G. Collee, J. P. Duguid, A. G. Fraser and B. P. Marimon, 1989, vol. 13, pp. 100–120 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20689a |
| This journal is © The Royal Society of Chemistry 2016 |