
Aqueous synthesis and photochromic study of Mo/W oxide hollow microspheres†
Abstract
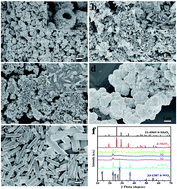
Mo/W oxide hollow microspheres with MoO3 as matrix material were fabricated via a polyethylene glycol (PEG) induced aqueous route. The hierarchical Mo/W oxide samples are constructed by elementary fusiform particles, which were characterized by X-ray powder diffraction (XRD) and scanning electron microscopy (SEM) methods. Initial formed h-WO3 crystallites acted as nuclei for subsequent formation of MoO3 precipitates, thus Mo/W oxide samples formed through epitaxial growth strategy show weak crystallization because of the lattice mismatch between h-MoO3 and h-WO3 crystals. The MoO3 based Mo/W oxide samples exhibit good photochromic performances compared to unitary h-MoO3 and h-WO3 crystals.
 Please wait while we load your content...
Please wait while we load your content...

