DOI:
10.1039/C6RA20650F
(Paper)
RSC Adv., 2016,
6, 102482-102497
Exploration of biological activities of alkyne arms containing Cu(II) and Ni(II) complexes: syntheses, crystal structures and DFT calculations†
Received
16th August 2016
, Accepted 21st October 2016
First published on 21st October 2016
Abstract
Several mononuclear complexes [Cu(L1), Cu(L2), Ni(L1) and Ni(L2)] containing N,O-donor Schiff base [L1 = 2,2′-{cyclohexane-1,2-diylbis[nitrilo(E)methylylidene]}bis[5-(prop-2-yn-1-yloxy)phenol] and L2 = 2,2′-{1,2-phenylenebis[nitrilo(E)methyllidene]}bis[5-(prop-2-yn-1-yloxy)phenol]] have been synthesized and characterized by elemental analyses and spectral techniques. The solid-state structures of Cu(L1) and Ni(L1) were determined using single-crystal X-ray crystallography reveal distorted square planar geometry around the metal ions. The binding ability of the complexes with DNA and BSA were investigated. DNA cleavage activity reveals that the complexes cleaved the plasmid DNA via hydrolytic path way. In vitro cytotoxicity assays indicate that these complexes exhibit anticancer activity against human cervical carcinoma cell line (HeLa). Flow cytometric analysis and Annexin V/PI double staining assays suggest that these complexes induce cell apoptosis. The comet assay has been employed to find out the extent of DNA fragmentation in cancer cells. The catecholase activity of the complexes has been investigated by UV-vis, cyclic voltammetry and EPR using 3,5-DTBC as model substrate reflect that the complexes are effective in mimicking catecholase like activity. DFT calculation on the mechanistic insights of catalytic activity suggests a ligand-centred radical mechanism.
Introduction
Terminal alkynes are extensively used in organic synthesis, pharmaceutical science, material science and bio-orthogonal chemistry. Several natural products contain a terminal alkyne, which gives the impression that an acetylenic functionality is an essential component for their bioactivity.1 Cancer is one of the most dreadful and serious diseases, which has significant influence on public health and causes death in humans. Synthesis of novel chemotherapeutic agents with high efficiency and low toxicity is a great challenge in the development of anticancer drugs. The primary target molecule for most anticancer drugs is DNA, and they exert their effect by interfering with DNA processing leading to cell death through apoptosis. Hence, research on the mechanism of binding modes of transition metal complexes with DNA have long been a thrust area and basic work of both inorganic and biochemists. Some platinum based drugs show potential efficacy in treating different types of cancers,2 but they show several side effects. So novel non-platinum-based chemotherapeutics drugs are developed as choice of platinum based drugs, which involved non-covalent modes of interaction with DNA.3 Among the various bio-essential metal complexes, copper(II) and nickel(II) complexes are considered as better cancer inhibiting activity.4 The design of copper(II) and nickel(II) complexes, which can bind with specificity to DNA and carry out its cleavage, has become essential in the development of novel antitumor agents.5 The development of metal complexes which cleave nucleic acids without any external agent under mild condition, is attracted in the field of artificial metallonucleases.6 Several nickel complexes have been examined due to their good DNA binding and cleavage activity.7 In addition, several copper(II) and nickel(II) complexes activate apoptotic cell death as a result of DNA damage occurring inside the cell by triggered pro-apoptotic proteins or anti-apoptotic proteins.4,8 The interaction of metal complexes with proteins becomes vital because it gives valuable information on the structural topography to find out the therapeutic effectiveness of drugs. Bovine serum albumin (BSA) has been examined widely for its structural homology with human serum albumin (HSA).9 Therefore, it is significantly important to investigate the interaction between metal complexes and BSA.
Catecholase activity of some model transition metal complexes has been a research area for the development of novel bio-catalysts.10 The oxidation of o-diphenols to corresponding quinone in presence of the enzyme catecholase is known as catecholase activity.11 The resulting quinone obtained is a highly reactive compound and can undergo auto-polymerization to give brown pigments (melanin). These brown pigments are responsible to defense the tissues from damage against pathogens and insects.12 As the enzyme catecholase has dimeric-copper active center, most of the research have been focused on di-nuclear copper(II) and nickel(II) complexes to study the possible catechol oxidase like properties. Only few reports are available where mononuclear copper and nickel complexes have been studied for possible catecholase like activities.
We have been working on the interaction of Schiff base metal complexes with DNA and protein as well as their anticancer activity.13 In the present work we are reporting the synthesis and characterization of four new mononuclear complexes [Cu(L1), Cu(L2), Ni(L1) and Ni(L2)] that contains an N,O-donor alkyne arms Schiff bases [L1 = 2,2′-{cyclohexane-1,2-diylbis[nitrilo(E)methylylidene]}bis[5-(prop-2-yn-1-yloxy)phenol] and L2 = 2,2′-{benzene-1,2-diylbis[nitrilo(E)methylylidene]}bis[5-(prop-2-yn-1-yloxy)phenol]. The binding ability of the complexes with DNA and BSA has been studied. The MTT assay and Annexin V/PI double staining assay was used to evaluate the in vitro cytotoxic activity and cell apoptosis against HeLa cell lines. The catecholase activity of all the complexes have been investigated using 3,5-DTBC as model substrate and the DFT calculations further have been done to suggest the probable mechanistic pathway involved in that activity.
Results and discussion
Synthesis and stability
The Schiff bases (L1 and L2) were prepared by condensing 2-hydroxy-4-(prop-2-yn-1-yloxy)benzaldehyde with 1,2-diaminocyclohexane/o-phenylenediamine in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in dehydrated alcohol. The lavender colored copper, and brick red nickel complexes were obtained by the reaction of L1 and L2 with Cu(CH3COO)2·H2O and Ni(CH3COO)2·H2O respectively in dehydrated methanol (Scheme 1). To find the stability of the compounds in solvents, each sample was incubated at 37 °C for 0, 8, 24, and 48 h, respectively and the UV-visible spectra were measured in the range of 250–800 nm at room temperature. Comparison of UV-visible spectra of the compounds in solvents or in buffer at room temperature revealed that no significant hydrolysis could be observed after incubation suggests that the compounds are stable in these solvents (Fig. S1†).
:1 molar ratio in dehydrated alcohol. The lavender colored copper, and brick red nickel complexes were obtained by the reaction of L1 and L2 with Cu(CH3COO)2·H2O and Ni(CH3COO)2·H2O respectively in dehydrated methanol (Scheme 1). To find the stability of the compounds in solvents, each sample was incubated at 37 °C for 0, 8, 24, and 48 h, respectively and the UV-visible spectra were measured in the range of 250–800 nm at room temperature. Comparison of UV-visible spectra of the compounds in solvents or in buffer at room temperature revealed that no significant hydrolysis could be observed after incubation suggests that the compounds are stable in these solvents (Fig. S1†).
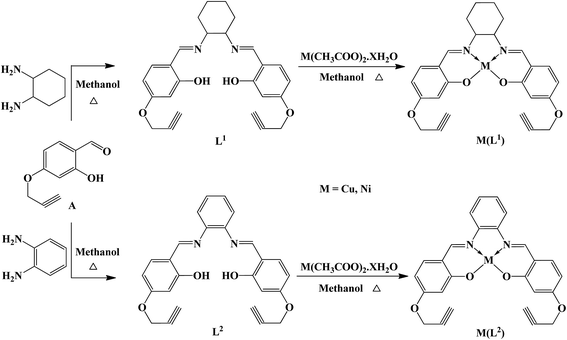 |
| | Scheme 1 Schematic presentation of preparation of Schiff bases and their metal complexes. | |
Spectroscopy
IR spectra of L1 and L2 exhibit broad band around 3465–3385 cm−1 are due to the presence of O–H⋯N intramolecular hydrogen bond (Fig. S2†).14 The terminal alkyne exhibits ν–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H and (ν–CC–) bands in the region 3284–3279 cm−1 and 2168–2162 cm−1 respectively in both the free ligands and metal complexes. The C
C–H and (ν–CC–) bands in the region 3284–3279 cm−1 and 2168–2162 cm−1 respectively in both the free ligands and metal complexes. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching frequency appeared at 1639 cm−1 (L1) and at 1631 cm−1 (L2) was shifted to a lower values in the complexes indicating the coordination of azomethine N to metal ions. The electronic absorption spectra of L1 and L2 show bands around 256 nm and 262 nm correspond to π → π* transitions, whereas the band around 340 nm and 349 nm is due to n → π* transitions (Fig. S3a†). The absorption spectra of the complexes show intra-ligand transitions. The broad band appeared in the region of 575–582 nm for copper complexes [1A1g → 1B1g], and 541–543 nm for nickel complexes [1A1g → 1B2g] corresponds to d–d transitions (Fig. S3b†). In the 1H NMR spectra of nickel complexes, the –OH signal registered in the range 12–14 ppm for the Schiff bases were disappeared representing deprotonation of the hydroxyl group due to Ni–O bond formation. The azomethine proton (N–CH–) was shifted to higher region upon the coordination of –Ni–NC– clearly demonstrating the binding of metal ion with the Schiff base through deprotonated –OH and azomethine N atoms (Fig. S4†). The EPR spectra of the copper complexes at room temperature consist of a weak shoulder at g∥ 2.21 for Cu(L1) and 2.35 for Cu(L2) and an intense signal at g⊥ 2.06 for Cu(L1) and 2.12 for Cu(L2) having a typical derivative line shape. These are well-known features in the EPR spectra of mononuclear Cu(II) complexes (S = 1/2). The observed g values (g∥ > g⊥ > 2.00) in both the complexes suggest that the unpaired electron in the Cu(II) ion is in the dx2−y2 orbital,15 and therefore, a square based geometry around the copper(II) is expected in both the complexes. The hyperfine structure has been observed only on the parallel component due to the interaction of unpaired electrons of Cu(II) with 63,65Cu having nuclear spin I = 3/2.16 To minimize the broadening of the signal in the complexes and to determine the hyperfine interactions and super-hyperfine interactions if any, the EPR spectra were recorded in acetonitrile at 77 K (Fig. S5†). The g∥ value of 2.13 for Cu(L1) and 2.11 for Cu(L2) complexes indicates the covalent nature of the metal–ligand bond. For the present square planar complexes, G = 2.10 and 2.22 indicate that the Schiff base is in strong field and the metal–Schiff base bonding in the complex is covalent.17 The bonding parameters α2 (in-plane σ bonding), β2 (in-plane π bonding) and γ2 (out of plane π bonding) were calculated demonstrate that there is an interaction in the in-plane π bonding between the metal ion and the Schiff base. This is also confirmed by orbital reduction factors, K∥ and K⊥ (Table S1†).
N stretching frequency appeared at 1639 cm−1 (L1) and at 1631 cm−1 (L2) was shifted to a lower values in the complexes indicating the coordination of azomethine N to metal ions. The electronic absorption spectra of L1 and L2 show bands around 256 nm and 262 nm correspond to π → π* transitions, whereas the band around 340 nm and 349 nm is due to n → π* transitions (Fig. S3a†). The absorption spectra of the complexes show intra-ligand transitions. The broad band appeared in the region of 575–582 nm for copper complexes [1A1g → 1B1g], and 541–543 nm for nickel complexes [1A1g → 1B2g] corresponds to d–d transitions (Fig. S3b†). In the 1H NMR spectra of nickel complexes, the –OH signal registered in the range 12–14 ppm for the Schiff bases were disappeared representing deprotonation of the hydroxyl group due to Ni–O bond formation. The azomethine proton (N–CH–) was shifted to higher region upon the coordination of –Ni–NC– clearly demonstrating the binding of metal ion with the Schiff base through deprotonated –OH and azomethine N atoms (Fig. S4†). The EPR spectra of the copper complexes at room temperature consist of a weak shoulder at g∥ 2.21 for Cu(L1) and 2.35 for Cu(L2) and an intense signal at g⊥ 2.06 for Cu(L1) and 2.12 for Cu(L2) having a typical derivative line shape. These are well-known features in the EPR spectra of mononuclear Cu(II) complexes (S = 1/2). The observed g values (g∥ > g⊥ > 2.00) in both the complexes suggest that the unpaired electron in the Cu(II) ion is in the dx2−y2 orbital,15 and therefore, a square based geometry around the copper(II) is expected in both the complexes. The hyperfine structure has been observed only on the parallel component due to the interaction of unpaired electrons of Cu(II) with 63,65Cu having nuclear spin I = 3/2.16 To minimize the broadening of the signal in the complexes and to determine the hyperfine interactions and super-hyperfine interactions if any, the EPR spectra were recorded in acetonitrile at 77 K (Fig. S5†). The g∥ value of 2.13 for Cu(L1) and 2.11 for Cu(L2) complexes indicates the covalent nature of the metal–ligand bond. For the present square planar complexes, G = 2.10 and 2.22 indicate that the Schiff base is in strong field and the metal–Schiff base bonding in the complex is covalent.17 The bonding parameters α2 (in-plane σ bonding), β2 (in-plane π bonding) and γ2 (out of plane π bonding) were calculated demonstrate that there is an interaction in the in-plane π bonding between the metal ion and the Schiff base. This is also confirmed by orbital reduction factors, K∥ and K⊥ (Table S1†).
Description of crystal structures
Thermal ellipsoid plots with the atomic labelling, crystal data and selected inter atomic bond lengths and angles of Cu(L1) and Ni(L1) complexes are shown in Fig. 1 and Tables 1, S2 and S3.† The complexes crystallized in monoclinic P21/n space group with Z of 4. The cyclohexyl ring of the complexes adopts a chair conformation with the two imine groups linked at equatorial positions. The crystal structure of Cu(L1) and Ni(L1) confirm a tetradentate N2O2 coordination of the Schiff base with the metal ions. The coordination environment of the metal ion is fulfilled by the two phenoxo oxygen and two imine nitrogen atoms of the Schiff base. The complex Cu(L1) has adopted a slightly distorted square planar geometry with the O(3)–Cu(1)–N(2), O(2)–Cu(1)–N(1) bond angles smaller than 180° measuring 174.92(12) and 175.65(11) degrees. The Cu–N bond distances are nearly identical and slightly longer than the Cu–O bond distances [Cu(1)–N(2) 1.923(3) Å, Cu(1)–N(1) 1.927(3) Å and Cu(1)–O(2) 1.887(2) Å, Cu(1)–O(3) 1.888(2)]. The central chelate ring Cu(1)–N(1)–C(1)–C(2)–N(2) and Cu(1)–N(2)–C(2)–C(1)–N(1) is an envelope on C(1) and C(2). Such a conformation results from the twist of the cyclohexyl moiety relative to the CuN2O2 coordination plane caused by the chirality of centers C(1) and C(2). The cyclohexyl ring is found in the chair conformation with the alternating values of 55 and −55° for the ring torsion angles. The values are comparable with the structures of substituted Cu(salen) complexes reported earlier.18,19a The bond angles O(1)–Ni(1)–N(1) and O(2)–Ni(1)–N(2) smaller than 180° measuring 178.12(9) and 178.25(9) degrees, reveal that Ni(L1) complex has adopted a slightly distorted square planar geometry. The Ni–N bond length are nearly equal and are slightly longer than the Ni–O bond distances [Ni(1)–N(1) 1.850(2) Å, Ni(1)–N(2) 1.854(2) Å and Ni(1)–O(1) 1.8416(17)Å, Ni(1)–O(2) 1.8314(18)]. The central chelate ring Ni(1)–N(2)–C(11)–C(16)–N(1) and Ni(1)–N(1)–C(11)–C(16)–N(1) is an cover on C(11) and C(16). The cyclohexane ring adopts a chair conformation with two imines associated at equatorial positions and the ring torsion angle values are 57 and −57°. The CuN2O2 coordination plane are caused by the chirality center of C(11) and C(16) atoms. All the values are comparable with the structures of substituted Ni(salen) complexes reported earlier.18,19
 |
| | Fig. 1 ORTEP view of, (a) Cu(L1) (CCDC 1432869†) (b) Ni(L1) (CCDC 1432870†) (thermal ellipsoids are shown at 50% probability). | |
Table 1 Crystal data and structure refinement parameters for Cu(L1) and Ni(L1)
| Empirical formula |
C26H24Cu·N2O4 |
C26H24N2NiO4 |
| Formula weight |
492.01 |
487.18 |
| T [K] |
296(2) |
296(2) |
| λ [Å] |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
| a [Å] |
10.4808(12) |
7.8251(5) |
| b [Å] |
9.5955(8) |
19.6656(13) |
| c [Å] |
22.932(2) |
14.6693(11) |
| α [°] |
90 |
90 |
| β [°] |
93.828(5) |
100.416(3) |
| γ [°] |
90 |
90 |
| V [A3] |
2301.1(4) |
2220.2(3) |
| Z |
4 |
4 |
| ρ [Mg m−3] |
1.420 |
1.458 |
| Absorption coefficient [mm−1] |
0.984 |
0.910 |
| F(000) |
1020 |
1016 |
| Crystal size [mm] |
0.35 × 0.30 × 0.20 |
0.35 × 0.30 × 0.30 |
| θ range for data collection [°] |
1.78 to 28.35 |
1.75 to 28.47 |
| Index ranges [h, k, l] |
−13, 13; −12, 8; −30, 30 |
−6, 10; −24, 26; −19, 19 |
| Reflections collected |
18290 |
17545 |
| Independent reflections |
5613 |
5553 |
| R(int) |
0.0431 |
0.0396 |
| Data/restraints/parameters |
5613/0/298 |
5553/0/298 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0483, wR2 = 0.1318 |
R1 = 0.0427, wR2 = 0.1153 |
| R indices (all data) |
R1 = 0.0960, wR2 = 0.1675 |
R1 = 0.0686, wR2 = 0.1387 |
| Largest diff. peak/hole [e A−3] |
0.812 and −0.912 |
0.681 and −0.717 |
| Go fit on F2 |
1.007 |
1.084 |
The packing diagram of polymeric structure of 1 and 2 exhibits intermolecular C–H⋯O hydrogen bond (Table 2). In complex 1, the intermolecular C–H⋯O hydrogen bonding involves an –CH group of chair form of the cyclohexane moiety and the phenyl oxygen atom, O3 linked with the dimeric building unit and the same continues along the polymeric chain. The intermolecular C–H⋯O hydrogen bond shows that, each layer is associated to adjacent layers through two different types of hydrogen bonding, (a) coordinated phenyl O(2) and the alkyne C(24) of the ligand and (b) coordinated phenyl O(3) and hydrogen (H24) of the alkyne group, are responsible for the formation of 2D layer arrangements (Fig. 2a and S6a†). In complex 2, the structure shows the existence of intermolecular hydrogen bonding between the adjacent layers involving hydrogen (H26) of alkyne group in one layer and the metal coordinated phenyl O1 in the next layer with H(26)⋯(O1) = 2.409 Å. Also, alkyne carbon atom bonding with carbon atom of phenyl ring (C21⋯C21 = 3.340 Å) and oxygen atom (C1⋯O4 = 3.030 Å) are responsible for the formation of 2D layer arrangements (Fig. 2b and S6b†).
Table 2 Hydrogen bonding parameters for Cu(L1) and Ni(L1), D = donor, A = acceptor (Å, °)
| D–H⋯A |
D–H |
D⋯A |
H⋯A |
Angle |
| 1/2 − x, 1/2 + y, 1/2 − z. 1 − x, 1 − y, 1 − z. x, y, z. 1 − x, 1 − y, 1 − z. −1/2 + x, 1/2 − y, 1/2 + z. −1/2 + x, 1/2 − y, −1/2 + z. 2 − x, 1 − y, 1 − z. 1 − x, 1 − y, 1 − z. 1.5 − x, 1/2 + y, 1/2 − z. x, y, z. |
| Cu(L1) |
| C10–H10⋯C20 |
0.930 |
3.562 |
2.659 |
163.98 |
| C10–H10⋯C21a |
0.930 |
3.751 |
2.847 |
164.35 |
| C24–H24⋯O2 |
0.930 |
3.219 |
2.326 |
160.81 |
| C24–H24⋯O3b |
0.930 |
3.201 |
2.583 |
127.29 |
| C7–O1⋯C25 |
1.363 |
3.881 |
3.137 |
128.44 |
| C7–C25⋯O1c |
0.930 |
3.137 |
2.310 |
147.89 |
| C12–O2⋯C24 |
1.300 |
3.985 |
3.219 |
120.71 |
| C19–O3⋯C24d |
1.310 |
3.914 |
3.201 |
113.58 |
| C1–H1⋯O3 |
0.980 |
3.330 |
2.528 |
138.93 |
| C12–O2⋯C17e |
1.300 |
3.442 |
3.176 |
90.51 |
|
| Ni(L1) |
| C26–H26⋯O1f |
0.929 |
3.318 |
2.409 |
166.32 |
| C21–O4⋯C3g |
1.372 |
3.682 |
3.110 |
103.58 |
| C21–O4⋯C1h |
1.372 |
3.340 |
3.030 |
90.63 |
| C16–H16⋯O1f |
0.980 |
3.382 |
2.494 |
150.57 |
| C8–O1⋯H16i |
1.311 |
2.982 |
2.494 |
98.36 |
| C8–O1⋯H26 |
1.311 |
3.244 |
2.409 |
118.41 |
| C5–H5⋯C14f |
0.930 |
3.672 |
2.886 |
143.02 |
| C4–C5⋯H24Bj |
1.395 |
2.916 |
2.802 |
80.49 |
 |
| | Fig. 2 Crystal packing diagram of, (a) Cu(L1) and (b) Ni(L1) along the a-axis. | |
DNA binding studies
Absorption spectral studies. UV-vis spectral data of the Schiff bases and their metal complexes in Tris–HCl buffer in the absence and presence of CT-DNA exhibit hypochromism accompanied by bathochromic shift (Fig. S7†). In general, hyperchromism and hypochromism are the spectral features of DNA concerning the changes of its double helix structure.20 The interaction of Cu(L1) and Ni(L1) with CT-DNA was monitored by the hypochromism (∼8% decrease) of the band at λ = 292 and 274 nm (Fig. S7a and b†). For Cu(L2) and Ni(L2), the interaction with CT-DNA was monitored by the hypochromism (∼10% decrease) of the band at λ = 289 and 275 nm (Fig. S7b and c†). This hypochromic effect is due to the stacking interaction between the aromatic chromophore of the metal complexes and the base pairs of DNA.21 The absorption at λ = 270–300 nm in the spectra of the metal complexes experiences a blue shift after the addition of CT-DNA (Table S4†). The binding affinity of the compounds with DNA was studied from the spectroscopic titrations of the synthesized compounds in Tris–HCl buffer with increasing concentrations of CT-DNA (Fig. 3 and S8†). The intrinsic binding constant (Kb) was determined by using equation,
where [DNA] is the concentration of DNA in base pairs, the apparent absorption coefficient εa, εf and εb correspond to Aobs/[compound], the extinction coefficient of the free compound and the compound when fully bound to DNA. The plot of [DNA]/(εa − εf) vs. [DNA] gave a slope and the intercept which are equal to 1/(εb − εf) and 1/Kb (εb − εf), respectively; Kb is the ratio of the slope to the intercept. As listed in Table 3, the binding constants values are in the range of 1.8–5.38 × 104 M−1. All the complexes exhibit almost similar binding affinity towards DNA, but greater than that of Schiff bases. The Kb values determined differ by two order of magnitude with the classical intercalators (EB–DNA, 7.0 × 107 M−1).22a The observed Kb values indicate that all the metal complexes efficiently interact with the DNA and the binding order is 104 M−1. These observations are similar to that of previously reported complexes.19,22b,c The binding mode between metal complexes and DNA cannot be completely ascertained by the absorption spectroscopic studies. Additional experiments are required to further explain the binding mode.
 |
| | Fig. 3 Electronic absorption spectrum of Cu(L1) complex in Tris–HCl buffer upon addition of CT-DNA (arrow indicates that the absorption intensities decrease upon increasing DNA concentration) and plot of [DNA]/(εa − εf) versus [DNA] for the Schiff bases and their metal complexes with CT-DNA. | |
Table 3 DNA binding constant (Kb), Stern–Volmer constant (Kq) and the apparent binding constant (Kapp) for ligands and their metal complexes
| Compounds |
Kb (M−1) |
Kq (M−1) |
Kapp (M−1) |
| L1 |
1.80 × 104 |
1.98 × 104 |
6.22 × 104 |
| L2 |
1.91 × 104 |
2.11 × 104 |
6.49 × 104 |
| Cu(L1) |
4.88 × 104 |
4.26 × 104 |
7.93 × 104 |
| Ni(L1) |
4.26 × 104 |
3.92 × 104 |
7.17 × 104 |
| Cu(L2) |
5.98 × 104 |
3.51 × 104 |
6.95 × 104 |
| Ni(L2) |
5.38 × 104 |
4.02 × 104 |
7.71 × 104 |
Fluorescence spectral studies. Ethidium bromide (EB) is one of the most sensitive fluorescent probes that bind to DNA through intercalation mode.23,24 The decrease of fluorescence intensity of EB–DNA complex on the addition of metal complex may be due to the displacement of EB from a DNA sequence and/or by accepting the excited state electron of EB through a photo-electron transfer mechanism.25 Upon addition of increasing the concentration of the metal complex, the fluorescence intensity of the EB–DNA decreased at 605 nm (hypochromism) demonstrates the strength of binding of metal complex with CT-DNA.26 This quenching is due to accepting the excited state electron of EB through a photo-electron transfer mechanism. Further, the quenching data were analyzed by classical Stern–Volmer equation,
where F0 and F represent the emission intensities of EB–DNA in the absence and presence of the quencher, Kq is the quenching constant and [Q] is the quencher concentration. In the Stern–Volmer plots of F0/F versus [Q], the quenching constant (Kq) is given by the ratio of the slope to intercept. The Kq values for the ligands and the metal complexes are given in the Table 3. Further, the apparent binding constant (Kapp) was calculated using the equation,
where [complex] is the concentration of the compound at which there is 50% reduction in the fluorescence intensity of EB, KEB = 1.0 × 107 M−1 and [EB] = 2.5 μM.27 The binding constants and quenching constants of the Schiff bases and metal complexes suggest that the interaction of all the compounds with DNA via weak intercalative binding mode23 and also the metal complexes bind to CT-DNA more strongly than the Schiff bases. The typical fluorescence quenching curves of EB bound to DNA in the presence of synthesized compounds and Stern–Volmer plot of fluorescence titrations are shown in Fig. 4 and S9.†
 |
| | Fig. 4 Fluorescence quenching curves of EB bound to DNA in the presence of Cu(L1) complex (arrow indicates that the absorption intensities decrease upon increasing DNA concentration) and Stern–Volmer plot of the Schiff bases and their metal complexes with CT DNA. | |
Time-resolved emission studies. To ascertain the binding nature between the metal complex and DNA, time resolved emission studies were carried out. Ethidium bromide (EB) is a well known DNA binder that intercalates with little sequence selectivity. The emission life time of free EB and EB–DNA is 1.8 ns and 23 ns, respectively. The addition of small organic molecules or metal complexes capable of replacing EB from EB–DNA complex, leads to the reduction of emission life time of EB–DNA.28 The decay time in time-resolved fluorescence studies of the complexed fluorophore molecules decreases due to the loss of energy from the fluorophore molecules participating in collision with quencher molecules, resulting in reduced emission lifetime.29 The emission lifetime of free EB–DNA complex and the various concentrations of the metal complexes interact with DNA is depicted in Fig. S10† and the results are given in Table S5.† The average emission lifetime for tri-exponential iterative fitting is calculated based on the decay time and pre-exponential factors (α) using the following equation,30
| 〈τ〉 = (α1τ12 + α2τ22 + α3τ32)/(α1τ1 + α2τ2 + α3τ3) |
where τ is lifetime and α is fractional amplitude. The emission decay of EB–DNA complex was obtained with the lifetime of 20.55 ns. The small difference in emission life time of EB–DNA and metal complex bound EB–DNA suggests that there is electron transfer between the EB–DNA and metal complex.31 But, the reduction in emission life value of 2.45 ns shows that, its not possible to completely ruled out the intercalation between the metal complex and DNA.32
Viscosity measurement. To explain the nature of binding interaction between the metal complexes and DNA, the viscosity measurements of CT-DNA was carried out after treatment with different concentrations of metal complexes. The intercalation binding mode of compounds with DNA results in lengthening of DNA helix and hence enhances the relative specific viscosity of DNA.33 In contrast, partial or non-classical intercalation of compounds may bends (or kinks) the DNA helix, thus reducing its effective length and consequently its viscosity.34 The relative specific viscosities values of DNA in the absence and presence of the complexes were plotted against [complex]/[DNA]. The relative viscosities of DNA bound to ligands and its metal complexes slightly enhanced with increasing complex concentration suggests that the metal complex binds with DNA via weak intercalative mode (Fig. S11†).35
Protein binding studies. Electronic absorption titration of protein (BSA) with Schiff bases and their metal complexes were done to predict the type of quenching process. Upon the additions of the test compounds to BSA, lead to an increase in absorption intensity without affecting the position of absorption band (Fig. S12†). These results show that the type of interaction between compounds and BSA was a static quenching process.36 The fluorescence emission spectra and Stern–Volmer plot of BSA after the addition of the compounds are shown in the Fig. 5 and S13.† Addition of the test compounds to the solution of BSA resulted in a significant decrease of the fluorescence intensity observed at 337 nm and no appreciable shift after the addition of the samples. The observed hypochromicity has revealed that the compounds interact hydrophobically with the BSA protein.37 The fluorescence quenching is described by the Stern–Volmer relation,
| F0/F = 1 + Kqτ0[Q] = 1 + Ksv[Q] |
where F0 and F are represents the fluorescence intensities in the absence and presence of quencher. Kq is the quenching rate constant of protein, τ0 is the average lifetime of protein without quencher and [Q] is the concentration of the quencher.38 Ksv is a linear Stern–Volmer quenching constant. The quenching constant (Kq) can be calculated using the plot of (F0/F) versus log[Q]. When small molecules bind independently to a set of equivalent site, on a macromolecule, the equilibrium between free and bound molecules is demonstrated by the Scatchard equation,39
| log[(F0 – F)/I] = logKb + nlog[Q] |
where Kb is the binding constant of the complexes with BSA and n is the number of binding sites. From the plot of log[(F0 – F)/F] versus log[Q], the binding constant (Kb) values and the number of binding sites (n) have been obtained (Fig. S14†). The quenching constant (Kq), binding constant (Kb) and number of binding sites (n) for the interaction of the ligand and complexes with BSA are shown in Table 4. From the results, Schiff bases and their complexes have only one binding site available to interact with BSA. Results showed that complexes interact strongly with BSA compared to free ligands.
 |
| | Fig. 5 Fluorescence quenching spectrum of BSA in the presence of increasing amounts of Cu(L1) complex. [BSA] = 1 μM and [complex] = 0–50 μM and Stern–Volmer plot of the Schiff bases and their metal complexes with BSA. | |
Table 4 Protein binding constant (Kb), quenching constant (Kq) and number of binding sites (n) for ligands and their metal complexes
| Compounds |
Kb (M−1) |
Ksv (M−1) |
n |
| L1 |
5.37 × 105 |
4.88 × 104 |
0.92 |
| L2 |
5.97 × 105 |
5.61 × 104 |
0.97 |
| Cu(L1) |
4.28 × 106 |
3.81 × 105 |
1.01 |
| Ni(L1) |
4.89 × 106 |
3.34 × 105 |
1.09 |
| Cu(L2) |
5.12 × 106 |
3.87 × 105 |
1.11 |
| Ni(L2) |
5.36 × 106 |
4.09 × 105 |
1.19 |
Characteristics of synchronous fluorescence spectra
Synchronous fluorescence spectrum of BSA were measured before and after the addition of Schiff bases and their metal complexes to provide valuable information on the molecular microenvironment, particularly in the vicinity of the fluorophore functional groups.40 In BSA protein, the amino acid residues such as tyrosine, tryptophan and phenylalanine are responsible for the fluorescence property. The difference between the excitation and emission wavelength (Δλ = λem − λex) reflects the nature of the chromophore.41 The large Δλ value, such as 60 nm, is characteristic of tryptophan residue and a small Δλ value, such as 15 nm, is characteristic of tyrosine.42 The synchronous fluorescence spectra of BSA with different concentrations of the compounds were recorded at Δλ = 15 nm (Fig. S15†) and Δλ = 60 nm (Fig. S16†). Upon the addition of test compounds to the solution of BSA, the fluorescence intensity of tryptophan residue at 340 nm decreased. Similarly, the fluorescence intensity of tyrosine residue at 301 nm decreased by the addition of the compounds. The above results suggest that the interaction of complexes with BSA affects the conformation of both tryptophan and tyrosine micro-region.43
DNA cleavage activity and action mechanism
DNA cleavage is the process of the relaxation of the supercoiled circular conformation of plasmid DNA to nicked circular and/or linear conformation, which can be assessed by gel electrophoresis. The supercoiled form of pUC19 DNA (form I) might be cleaved by complexes with or without any external agents, thus causing them to relax to produce an open circular nicked form (form II).44 The cleavage of supercoiled pUC19 DNA by metal complexes (25 μM) was studied in a medium of 50 mM Tris–HCl/10 mM NaCl buffer (pH = 7.2) at 37 ± 0.2 °C. All the complexes exhibit notable DNA cleavage activity. Increase in concentration of the complexes promotes the relaxation of supercoiled DNA (form I, SC) to form a nicked circular DNA (form II, NC) and then the linear DNA (form III, LC) form (lanes 2–7) (Fig. 6 and S17–S19†).
 |
| | Fig. 6 Agarose gel showing cleavage of pUC19 DNA incubated by Cu(L1) in Tris–HCl buffer (pH = 7.2) at 37 °C for 1 h. Lane 1: DNA control; lane 2: 2.5 μM Cu(L1) + DNA; lane 3: 5.0 μM Cu(L1) + DNA; lane 4: 7.5 μM Cu(L1) + DNA; lane 5: 10.0 μM Cu(L1) + DNA; lane 6: 12.5 μM Cu(L1) + DNA; lane 7: 15.0 μM Cu(L1) + DNA. | |
The cleavage mechanism of pUC19 DNA provoked by complexes were examined and explained in the presence of singlet oxygen quenchers (L-histidine, 0.25 μM), superoxide scavenger (superoxide dismutase enzyme SOD, 4 units), hydroxyl radical scavengers (DMSO, KI, 0.1 mM), and chelating agent (EDTA, 5 mM) under aerobic conditions.45 The presence of scavengers (L-histidine, SOD, DMSO and KI) do not alter the DNA cleavage activity (Fig. S20†), and this rules out the cleavage of DNA by hydroxyl radical, singlet oxygen and superoxide anion. As a result, DNA cleavage promoted by the complexes takes place through a hydrolytic pathway. The chelating agent (EDTA, 5 mM) strongly binds with metal ion to form a stable complex, can effectively reduce the DNA cleavage activity, indicating that the added metal complexes play the vital role in the DNA cleavage (Fig. S21†). When the NC form obtained from the cleavage of SC DNA was reacted with a T4 ligase enzyme, complete conversion of NC DNA to its original SC form was observed (Fig. S22†). The results reveal that all the complexes performed as a synthetic nuclease and break the plasmid DNA in the absence of oxygen, probably through the hydrolytic mechanism.45
Cytotoxicity evaluation
Cytotoxicity of all the compounds was performed against HeLa cell line using the MTT assay (Fig. S23†). From the resulted values (Table S6†), all the compounds show a considerable inhibitory potency against proliferation of the cell line in a dose-dependent manner, with L1 and L2 as an exception having required a relatively higher concentration (IC50 = 19–23 μmol L−1). This suggests that the compounds have the ability to interact with the cell DNA, which is positively a valuable factor for allowing for these complexes as promising candidates for novel antitumor agents.
Annexin V/PI double staining
The mechanism of cell death caused by the metal complexes was determined using Annexin V-propidium iodide (PI) staining assay method. This staining assay clearly identified the viable, apoptotic or necrotic population resulting after the treatment of complexes. The HeLa cells were treated with each metal complex for a period of 48 h. The untreated cells remained completely viable with no sign of apoptosis or necrosis. All the four complexes exactly induced cancer cell apoptosis and mostly in the early phase (Fig. 7). These results are consistent with the MTT results. The results clearly show that the cellular death triggered by the metal complexes follow the apoptosis pathway as reported earlier.46 The fluorescence imaging method was used to visualize the effect of the metal complexes on the cancer cell (HeLa cell). After the treatment of metal complexes with the cancer cell, a bright field images were observed indicate the cell death with alteration in the cellular morphology. This includes shrinking of the cells followed by clustering and fragments formation (Fig. 8). This ascertained that the metal complexes mediated cell death has taken place due to apoptosis which was activated by fragmentation47 and damage of the DNA.
 |
| | Fig. 7 Apoptosis distribution of HeLa cells after treatment with 1–4 (10 μmol L−1) for 48 h. (a) Control untreated cells; (b) cells treated with [Cu(L1)]; (c) cells treated with [Ni(L1)]; (d) cells treated with [Cu(L2)]; (e) cells treated with [Ni(L2)]. I3: living cells, I4: early apoptotic cells (UR), I2: late apoptotic cells (LR) and I1: necrotic cells. | |
 |
| | Fig. 8 (A) Cell morphology on treatment with metal complexes. Panels (a)–(e) represent the control and treatment with the metal complexes [Cu(L1), Ni(L1), Cu(L2) and Ni(L2)]; (B) induced DNA damage in HeLa cell treated with complexes [Cu(L1), Ni(L1), Cu(L2) and Ni(L2) and images of fragmentation detected by the comet assay. | |
Comet assay
Single cell gel electrophoresis (comet assay) was used to determine whether the complexes induce DNA strand fragmentation, which is an indication of early apoptosis.48 As shown in Fig. 8, HeLa cells treated with the complexes show considerable well-formed comets, whereas the control (untreated) cells show a round shape. The result shows that the complexes significantly induce the DNA damage and the length of the comet tail characterizes the extent of DNA fragmentation, which is further support for induction of apoptosis by the complexes.
Catechol oxidase studies and kinetics
The catecholase-like activity of the metal complexes (10−4 M) were evaluated by observing the oxidation of 3,5-DTBC (3,5-di-tert-butylcatechol, 100 equiv.) by O2 to 3,5-DTBQ (3,5-di-tert-butylquinone) in acetonitrile medium. The reactions were carried out at 25 °C in aerobic condition and the course of the reaction was monitored by UV-vis spectroscopy at different time intervals. The spectra of complexes show drastic changes immediately after the addition of 3,5-DTBC at ∼400 nm due to the formation of the oxidized product 3,5-DTBQ (Fig. 9 and S24†). The formed product 3,5-DTBQ was purified by column chromatography using a hexane/ethyl acetate eluant mixture. The product was isolated by slow evaporation of the eluant and was characterized by determining its melting point (∼111 °C) which agreed well with that reported in literature.49 The compound was further identified by 1H, 13C-NMR and ESI mass spectroscopy (Fig. S25 and S26†). 1H NMR (CDCl3, 400 MHz): δ (ppm) = 1.26 (s, 9H), 1.41 (s, 9H), 6.74 (d, 1H), 6.89 (d, 1H). 13C NMR (CDCl3, 400 MHz): δ (ppm) = 29.70 (C10), 31.58 (C9), 34.35 (C5), 34.85 (C8), 110.47 (C3), 116.11 (C6), 138.79 (C7), 140.70 (4), 142.28 (C1) and 142.31 (C2).
 |
| | Fig. 9 Catecholase activity by change in time dependent spectral pattern of complexes [(a) Cu(L1); (b) Ni(L1)] after addition of 3,5-DTBC in acetonitrile medium. | |
The kinetics of oxidation of 3,5-DTBC by the metal complexes has been understood by monitoring the increased product (3,5-DTBQ) concentration. The rate constant for catalyst-complex mixture was determined from the log[Aα/(Aα − At)] versus time plot. In order to evaluate the dependence of the rates on the substrate concentration, the metal complexes were treated with different concentrations of 3,5-DTBC (10 to 100 equiv.) under air-saturated conditions. The first-order dependence on the substrate concentration was observed at low concentrations of 3,5-DTBC and saturation kinetics was found at higher concentrations of 3,5-DTBC. The observed rates versus concentration of substrate data were analyzed on the basis of the Michaelis–Menten approach of enzymatic kinetics to get the Lineweaver–Burk plot (Fig. 10 and S27†). The kcat order is Cu(L1) > Cu(L2) > Ni(L1) > Ni(L2) (Table 5). The kcat values obtained for complexes are appreciably higher than similar complexes reported in the literature (Table S7†). The probable catalytic cycle of oxidation of 3,5-DTBC by complexes were given in Schemes S1 and S2.†
 |
| | Fig. 10 Plot of rate vs. substrate concentration for (a) Cu(L1); (b) Lineweaver–Burk plot for Cu(L1); (c) Ni(L1); (d) Lineweaver–Burk plot for Ni(L1). | |
Table 5 Kinetic parameters for the oxidation of 3,5-DTBC catalyzed by complexes
| Catalyst |
Vmax (M s−1) |
Km (M) |
std error |
kcat (h−1) |
| Cu(L1) |
3.91 × 10−3 |
3.28 × 10−2 |
1.44 × 10−4 |
234.60 |
| Ni(L1) |
2.80 × 10−3 |
1.52 × 10−2 |
5.21 × 10−4 |
168.00 |
| Cu(L2) |
3.76 × 10−3 |
8.35 × 10−2 |
6.65 × 10−4 |
225.40 |
| Ni(L2) |
1.44 × 10−3 |
1.46 × 10−2 |
3.99 × 10−4 |
84.40 |
EPR titration
During the catalytic reaction, organic radical and/or intermediate species are produced by metal complexes. In order to identify the formation of free radicals during the course of the reaction, EPR spectra of the reaction mixture (10−3 M) containing each complex and 3,5-DTBC (10−1 M) in acetonitrile solution were taken at room temperature at different time intervals (within 30 min). Under the experimental conditions, the nickel salts and 3,5-DTBC are EPR-silent. This implies that oxidation of 3,5-DTBC is occurring via a radical pathway only when the metal complexes are used as catalysts. EPR results of metal complexes exhibit a weak signal at g ∼ 2 (free electron,50 g = 2.0023), which attributes the formation of an organic radical species as the reaction intermediate in the catalytic process (Fig. 11 and S28†).51
 |
| | Fig. 11 EPR spectra in different time intervals of acetonitrile solution of complexes [(a) Cu(L1); (b) Cu(L2)] after the addition of 3,5-DTBC at room temperature. | |
Electrochemical study
The electrochemical properties of copper and nickel complexes have been investigated by cyclic voltammetry in acetonitrile solution containing 0.1 M TBAP (tetra-butyl-ammonium perchlorate) as the supporting electrolyte in the potential range −1.2 to +1.2 V versus Ag/AgCl reference electrode at room temperature. An irreversible oxidation peak is observed at −0.12 V and 0.82 V (Epa) is probably due to Cu0/CuI and CuII/CuIII (Fig. S29a†). The quasi-reversible couple at 0.21 V (Epa) and −0.07 V (Epc) is due to the CuI/CuII and CuII/CuI. After the addition of 3,5-DTBC to the copper complex, the irreversible oxidative peak is replaced by a new peak at −0.28 V and 0.72 V. The quasi-reversible couple is replaced by a new couple at 0.16 V and −0.29 V. The results show that, the anodic peak at 0.16 V represent the oxidation of CuII bound 3,5-DTBC to free 3,5-DTBQ and the cathodic peak at −0.28 V represent the reduction of free 3,5-DTBQ to CuII bound deprotonated 3,5-DTBC. For Cu(L2), an irreversible oxidation peak at Epa values of 0.80 V is probably due to CuII/CuIII (Fig. S29b†). The quasi-reversible couple at −0.18 V, 0.21 V (Epa) and −0.26, −0.32 V (Epc) is due to the Cu0/CuI, CuI/CuII. After the addition of 3,5-DTBC, the irreversible oxidative wave is replaced by a −0.71 V (CuII/CuIII). The quasi-reversible couple is replaced by a new couple with −0.39, 0.18 V (Epa) and −0.29, −0.58 V (Epc). From the results, the anodic peak at 0.18 V is due to the oxidation of CuII bound 3,5-DTBC to free 3,5-DTBQ and the cathodic peak at −0.29 V due to the reduction of free 3,5-DTBQ to CuII bound deprotonated 3,5-DTBC.
All the NiII complexes show similar cyclic voltammograms with ligand-centered oxidation at 0.6 to 1.1 V in quasi-reversible nature (Fig. S29c–f†). The nickel complexes show one cathodic wave at ∼−1.10 V corresponds to the reduction of NiII/NiI. However, the complexes exhibits two anodic wave at ∼+0.68 and ∼+0.97 V. The former oxidation peak corresponds to the oxidation of NiII/NiIII and the latter one is due to the reduction of the –CN bond. After the addition of the complexes with 3,5-DTBC, the cathodic wave at ∼−1.05 V is shifted to ∼−1.10 V and the oxidation peaks at ∼+0.68, +0.97 V are shifted to ∼+0.61, +1.05 V. These results show that the variation in the oxidation state of metal complexes is due to the complex–3,5-DTBC aggregation.52
Electrospray ionization mass spectral study
To find a better considerate of the complex–substrate intermediate and a mechanistic inference of catecholase activity during the oxidation reaction, we have recorded electrospray ionization mass spectra (ESI-MS positive) of complexes and a 1:100 mixture of the complex and 3,5-DTBC within 5 min of mixing in acetonitrile medium. Electrospray mass spectra of L1 and L2 are given in Fig. S30a and b.† All the complexes exhibits a base peak at m/z = 493.10, 488.20, 486.85 and 481.16 (100%), which can be assigned to the “ligand complex” formation (Table S8 and Fig. S31a–S34a†). After the addition of 3,5-DTBC to the metal complexes, drastic changes are observed in the spectra and the peaks at m/z = 221.21 and 243.22 for [Cu(L1)], 221.35 and 243.29 for [Ni(L1)], 221.16 and 243.19 for [Cu(L2)], 221.28 and 243.30 for [Ni(L2)] can be assigned to the protonated quinone [(3,5-DTBQ)]H+ and the quinone–sodium aggregate [(3,5-DTBQ)]Na+, respectively. The peaks at m/z = 714.22, 731.12, 730.09 and 703.05 corresponding to the monocationic species [ML1(3,5-DTBC)]H+ of the metal complexes [Cu(L1), Ni(L1), Cu(L2) and Ni(L2)] (Fig. S31b–S34b†). These results reveal the formation of the catalyst–substrate as intermediates, which take part in substrate activation during the oxidation of 3,5-DTBC to 3,5-DTBQ in presence of oxygen. After the quinone molecule is released, the catalyst is regenerated and the catalytic cycle was continued. The oxygen that takes part in this process is converted to H2O2. The liberated H2O2 was identified and characterized spectrophotometrically.53 Hence it is concluded that in the catecholase reaction mechanism, electron transfer is mainly facilitated by metal center and then further delocalized via –CN bond of metal Schiff base complex to the adjacent conjugate system.
Detection of d–d transition band in the catalytic reactions
Time dependent absorption spectra of copper complexes were recorded in the range 500–900 nm after mixing of complex with 3,5-DTBC. After the addition, the intensity of the d–d band of copper complexes is decreased in 500–600 nm range (Fig. S35a and b†) indicates that electron transfer process from catechol to the copper(II) center, which is consequently reduced to copper(I).49 Similarly, time dependent UV-vis spectra of Ni(L1) and Ni(L2) complexes were recorded in the range 500–900 nm with 3,5-DTBC. After the addition, the d–d band intensity of nickel complexes is decreased (Fig. S35c and d†) indicates that that coordination of Ni(II) changes from 4 to 5 or 6 during development of complex–substrate aggregate.7
DFT calculations
In order to get a better understanding of nature of the active species formed during catalysis, we carried out DFT analysis at the B3LYP/6-311G (d,p) level using the Gaussian 09 software.54 Calculations have been done on the metal complexes on the corresponding one-electron-reduced analogues Cu(L1)−, Ni(L1)−, Cu(L2)− and Ni(L2)−. The one-electron-reduced monoanionic species display a doublet (S = 1/2) ground state and the changes in net spin density visibly show a ligand-centered process for the reductive reaction (Fig. 12). The result shows that, one unpaired electron mainly localized in the –CN bond and carbon atoms of the aromatic ring and the other electron delocalized in the catechol ring and oxygen atoms. Therefore, the proposed electron transfer mechanism is supported by the orbital distribution in complexes and the electron transfer is facilitated by the metal center. The imine bond, –CN in the monoanionic species is significantly elongated (∼0.029 Å), indicating the formation of imine radicals.51b The optimized structural data of all the complexes is given in Tables S9 and S10.†
 |
| | Fig. 12 Spin density plots of, (a) Cu(L1)−; (b) Ni(L1)−; (c) Cu(L2)− and (d) Ni(L2)−. | |
Conclusions
Four new Cu(II) and Ni(II) Schiff base complexes were synthesized and structurally characterized. The complexes have a slightly distorted square planar geometry with the coordination environment of the metal ion is fulfilled by two phenoxo oxygen and two imine nitrogen atoms of the Schiff base. DNA binding studies reveals that all the compounds interact with DNA and the metal complexes have stronger binding affinity to DNA than the Schiff bases. The time-resolved lifetime measurement clearly shows that all the metal complexes were effectively interact with CT-DNA. Viscosity studies suggest that metal complexes bind with DNA through weak intercalation mode. The protein binding studies suggest that all the complexes could bind BSA protein with high affinity and quench the fluorescence of BSA through a static quenching mechanism. The synchronous fluorescence measurement results reveal that all the complexes influence the microenvironment around both tyrosine and tryptophan residues in BSA. The results of DNA cleaving proved that all the complexes under hydrolytic condition behave as efficient DNA nuclease, considering that the supercoiled form evidently decreased and changed into other forms. The metal complexes demonstrate better cytotoxic activity against HeLa cells. Fluorescence staining techniques and flow cytometry using the Annexin-V assay reveal that all the complexes induce apoptosis in cancer cells. Thus, this work is of great importance towards the design and development of efficient Cu(II) and Ni(II) complex based artificial nucleases and for their potential applications as therapeutic agents. All the complexes display promising catecholase like activity, apart from the above revealed biomolecule interactions. The catalytic reaction of the complexes in acetonitrile follows first order reaction pathway. The EPR experiment clearly shows ligand-centered catechol oxidation by complexes. Cyclic voltammetric studies also strengthen these results, indicating the feasibility of the radical pathway in catecholase activity for these complexes. The experimental results are confirmed by DFT analyses that corroborate a ligand-centered radical in the reduced complexes with significant elongation of the imine bond length. In general this class of transition metal complexes have shown promising biological activities and further studies are underway with similar class of ligands.
Experimental section
Physical methods and materials
The melting points were determined on Lab India instrument and are uncorrected. The elemental analyses (carbon, hydrogen and nitrogen) were performed using a Thermo Finnigan Flash EA 1112 series CHNS analyzer. FT-IR spectra (4000–400 cm−1) were recorded at room temperature using a Shimadzu 8400S spectrophotometer. Electronic absorption spectra were recorded using a Shimadzu-2600 Spectrophotometer. Emission spectra were measured on a Jasco FP-8300 Spectrofluorometer. Time-resolved fluorescence lifetime measurements were recorded using Horiba JobinYvon spectrofluorometer.1H NMR and 13C NMR measurements were recorded in CDCl3 by using TMS as an internal standard on a Bruker Avance 400 MHz spectrometer. EPR (X-band, 9.78 GHz) spectra were recorded using a Bruker EMX Plus instrument. Mass spectrometric analyses were done on Bruker-Daltonics, microTOF-Q II mass spectrometer. Cyclic voltammetry studies for all the complexes (0.001 M) were performed on a CHI620E spectroelectrochemical workstation in acetonitrile solution containing 0.1 M tetrabutylammonium perchlorate (TBAP) as supporting electrolyte. The experiments were carried out in a conventional three-electrode cell composed of a reference electrode Ag/AgCl, a platinum wire as auxiliary electrode and a glassy carbon as working electrode. All the chemicals were obtained from commercial sources and used as received. Solvents were dried according to standard procedure and distilled prior to use. 2,4-Dihydroxy benzaldehyde, propargyl bromide, potassium bicarbonate, potassium iodide, trans-1,2-diaminocyclohexane, o-phenylenediamine, metal salts and 3,5-di-tert-butylcatechol (3,5-DTBC) were purchased from Aldrich.
Synthetic procedures
2,2′-{Cyclohexane-1,2-diylbis[nitrilo(E)methylylidene]}bis[5-(prop-2-yn-1-yloxy)phenol] (L1). The compound 2-hydroxy-4-(prop-2-yn-1-yloxy)benzaldehyde (A) was synthesized as reported in our earlier work.13a Schiff base (L1) was synthesized by the addition of 10 mL methanolic solution of trans-1,2-diaminocyclohexane (30 μL, 0.25 mmol) to A (88 mg, 0.5 mmol). The reaction mixture was refluxed for 36 h. After evaporating the volatile solvent a pale yellow solid was obtained. Yield: 80%. Pale yellow color solid. Mp: 191 °C. Anal. calcd for C26H26O4N2. Found (calculated) (%) C: 72.51 (72.54), H: 6.05 (6.09), N: 6.49 (6.51). UV-vis (CH3CN): λmax = 256, 340 nm. FT-IR (KBr): ν, cm−1 3465–3385 (O–H⋯N), 3284 (–CC–H), 2918 (asymmetric –C–H), 2848 (symmetric –C–H), 2168 (–CC–), 1639 (–CN). 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.36–1.98 (10H, m, CH2–cyclohexane ring), 3.81–3.82 (1H, d, N–CH–), 6.51–7.30 (6H, m, aryl), 2.53–2.55 (2H, s, –CCH), 4.70 (4H, s, –O–CH2), 8.49 (s, azomethine), 13.24 (s, –OH). 13C NMR (400 MHz, CDCl3): δ (ppm) = 161.81–163.99 (azomethine), 62.99 and 75.92 (C9 and C10), 27.29 (C22, C23), 33.61 (C21, C24), 77.92 (C1, C18), 55.88 (C3, C16), 102.36–133.61 (aromatic) (Fig. S36†).
2,2′-{Benzene-1,2-diylbis[nitrilo(E)methylylidene]}bis[5-(prop-2-yn-1-yloxy)phenol] (L2). The Schiff base (L2) was synthesized by the addition of 10 mL methanolic solution of o-phenylenediamine (27 mg, 0.25 mmol) to A (88 mg, 0.5 mmol). The reaction mixture was refluxed for 36 h. After evaporating the volatile solvent a yellow solid compound (L2) was formed. Yield: 89%. Yellow color solid. Mp: 198 °C. Anal. calcd for C26H20O4N2. Found (calculated) (%) C: 73.56 (73.57), H: 4.73 (4.75), N: 6.58 (6.60). UV-vis (CH3CN): λmax = 262, 349 nm. FT-IR (KBr): ν, cm−1 3465–3385 (O–H⋯N), 3279 (–CC–H), 2909 (asymmetric –C–H), 2844 (symmetric –C–H), 2162 (–CC–), 1631 (–CN). 1H NMR (400 MHz, CDCl3): δ (ppm) = 6.51–7.30 (6H, m, aryl), 2.56–2.57 (2H, s, –CCH), 4.74 (4H, s, –O–CH2), 8.54 (s, azomethine), 13.31 (s, –OH). 13C NMR (400 MHz, CDCl3): δ (ppm) = 161.78–163.85 (azomethine), 140.30 and 142.58 (C9 and C10), 77.88 (C1, C18), 55.81 (C3, C16), 102.36–113.89 (phenolic aromatic), 119.54–133.61 (aromatic, amine) (Fig. S36†).
Synthesis of Cu(L1). 10 mL of methanolic solution containing L1 (0.22 g, 0.5 mmol) and Cu(OAc)2·H2O (0.10 g, 0.5 mmol) was stirred at room temp for 1 h and resulting lavender colored solution was kept in the air for few days until the solvent evaporates to furnish a lavender colored solid. After that this dried compound was dissolved in DMSO and layered by methanol for crystallization. Dark magenta color block shaped crystal of Cu(L1) was obtained after one week time. Yield: 79.72%. Lavender color solid. Mp: 278 °C. Anal. calcd for C26H24CuN2O4. Found (calculated) (%) C: 63.44 (63.47), H: 4.89 (4.92), N: 5.65 (5.69). UV-vis (CH3CN): λmax = 251, 281, 347, 541 nm. FT-IR (KBr): ν, cm−1 3279 (–CC–H), 2910 (asymmetric –C–H), 2842 (symmetric –C–H), 2160 (–CC–), 1631 (–CN).
Synthesis of Cu(L2). 15 mL of methanolic solution containing L2 (0.21 g, 0.5 mmol) and Cu(OAc)2·H2O (0.10 g, 0.5 mmol) was stirred at room temp for 3 h and resulting brown colored solution was concentrated by evaporating the solvent. After evaporated brown colored solid of Cu(L2) was formed. Yield: 82.19%. Brown color solid. Mp: 288 °C. Anal. calcd for C26H18CuN2O4. Found (calculated) (%) C: 64.24 (64.26), H: 3.70 (3.73), N: 5.73 (5.76). UV-vis (CH3CN): λmax = 257, 290, 353, 543 nm. FT-IR (KBr): ν, cm−1 3269 (–CC–H), 2913 (asymmetric –C–H), 2846 (symmetric –C–H), 2161 (–CC–), 1623 (–CN).
Synthesis of Ni(L1). 10 mL of methanolic solution containing L1 (0.22 g, 0.5 mmol) and Ni(CH3COO)2·4H2O (0.12 g, 0.5 mmol) was stirred at room temp for 2 h and resulting brick red colored solution was evaporating to dryness. After evaporated brick red colored solid of Ni(L1) was formed. After that this dried compound was dissolved in DMSO and layered by methanol for crystallization. Brick red color block shaped crystal of Cu(L1) was obtained after one week time. Yield: 82.19%. Brick red color solid. Mp: 292 °C. Anal. calcd for C26H24NiN2O4. Found (calculated) (%) C: 64.07 (64.10), H: 4.96 (4.97), N: 5.73 (5.75). UV-vis (CH3CN): λmax = 232, 275, 386, 575 nm. FT-IR (KBr): ν, cm−1 3275 (–CC–H), 2910 (asymmetric –C–H), 2840 (symmetric –C–H), 2163 (–CC–), 1626 (–CN). 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.91 (–Ni–NC–).
Synthesis of Ni(L2). 15 mL of methanolic solution containing L2 (0.21 g, 0.5 mmol) and Ni(CH3COO)2·4H2O (0.12 g, 0.5 mmol) was stirred at room temp for 3 h and resulting brick red colored solution was evaporating to dryness. After evaporated brick red colored solid of Ni(L1) was formed. Yield: 85.51%. Brick red color solid. Mp: 296 °C. Anal. calcd for C26H18NiN2O4. Found (calculated) (%) C: 64.89 (64.91), H: 3.76 (3.77), N: 5.80 (5.82). UV-vis (CH3CN): λmax = 235, 312, 383, 582 nm. FT-IR (KBr): ν, cm−1 3272 (–CC–H), 2912 (asymmetric –C–H), 2845 (symmetric –C–H), 2159 (–CC–), 1630 (–CN). 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.62 (–Ni–NC–).
Single crystal X-ray diffraction studies
A Bruker Kappa Apex II X-ray diffractometer was used for crystal screening, unit cell determination and data collection. The X-ray radiation employed was generated from a Mo sealed X-ray tube (Kα = 0.70173 Å at 296(2) K with a potential of 40 kV, 40 mA) attached with a graphite monochromator in the parallel mode (175 mm collimator with 0.5 mm pinholes). The sixty data frames were taken at widths of 0.5°. These reflections were used in the auto-indexing method to determine the unit cell. The relevant cell was determined and refined by nonlinear least squares and Bravais lattice methods. The unit cell was confirmed by check the h k l overlays on various frames of data by comparing with both the orientation matrices. No super-cell or erroneous reflections were observed. The integrated intensity data for each reflection was collected by reduction of the of the data frames with the program Apex II.55 The geometrical parameters were obtained using PARST and SHELXL-97. Hydrogen atoms were located in perfect positions and were set riding on the individual parent atoms. All non-hydrogen atoms were refined with anisotropic thermal parameters. The structure was refined (full matrix least square method on F2) to convergence. Olex2 was used for the final data arrangement and structure plots.56
Acknowledgements
The financial supports received from the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India, New Delhi (EMR-II/2014/000081) and Board of Research in Nuclear Sciences (BRNS), DAE-BARC, Mumbai, India (No. 35/14/03/2014) is gratefully acknowledged. C. B. is thankful to CSIR, India for his fellowship. SAIF-STIC, Cochin is acknowledged for CHNS and SCXRD analysis.
References
-
(a) R. E. Minto and B. J. Blacklock, Prog. Lipid Res., 2008, 47, 233–306 CrossRef CAS PubMed
 ;
(b) M. Yamaguchi, H. J. Park, S. Ishizuka, K. Omata and M. Hirama, J. Med. Chem., 1995, 38, 5015–5022 CrossRef CAS PubMed .
;
(b) M. Yamaguchi, H. J. Park, S. Ishizuka, K. Omata and M. Hirama, J. Med. Chem., 1995, 38, 5015–5022 CrossRef CAS PubMed . -
(a) G. Annarita, A. Maurizio, C. Claudio, G. Elisabetta and O. Domenico, J. Inorg. Biochem., 2004, 98, 73–78 CrossRef ;
(b) E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed ;
(c) Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed ;
(d) E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed .
-
(a) C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed ;
(b) A. N. Kate, A. A. Kumbhar, A. A. Khan, P. V. Joshi and V. G. Puranik, Bioconjugate Chem., 2014, 25, 102–114 CrossRef CAS PubMed ;
(c) D. Gaynora and D. M. Griffith, Dalton Trans., 2012, 41, 13239–13257 RSC ;
(d) B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed .
-
(a) D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccis and B. Graham, Coord. Chem. Rev., 2012, 256, 897–937 CrossRef CAS ;
(b) L. Marcelis, J. Ghesquiere, K. Garnir, A. K. D. Mesmaeker and C. Moucheron, Coord. Chem. Rev., 2012, 256, 1569–1582 CrossRef CAS ;
(c) C. Marzano, M. Pellei, F. Tisano and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS .
-
(a) I. Kostova, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 591–602 CrossRef CAS PubMed ;
(b) G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. Garcla, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef CAS .
-
(a) J. He, P. Hu, Y. J. Wang, M. L. Tong, H. Sun, Z. W. Mao and L. N. Ji, Dalton Trans., 2008, 3207–3214 RSC ;
(b) J. L. G. Gimenez, M. G. Alvarez, M. L. Gonzalez, B. Macias, J. Borras and G. Alzuet, J. Inorg. Biochem., 2009, 103, 923–934 CrossRef PubMed ;
(c) P. Jaividhya, R. Dhivya, M. A. Akbarsha and M. Palaniandavar, J. Inorg. Biochem., 2012, 114, 94–105 CrossRef CAS PubMed .
- M. Das, R. Nasani, M. Saha, S. M. Mobin and S. Mukhopadhyay, Dalton Trans., 2015, 44, 2299–2310 RSC .
-
(a) A. Haleel, P. Arthi, N. D. Reddy, V. Veena, N. Sakthivel, Y. Arun, P. T. Perumal and A. K. Rahiman, RSC Adv., 2014, 4, 60816–60830 RSC ;
(b) P. Vijayan, P. Viswanathamurthi, K. Velmurugan, R. Nandhakumar, M. D. Balakumaran, P. T. Kalaichelvan and J. G. Malecki, RSC Adv., 2015, 5, 103321–103342 RSC .
-
(a) A. Sulkowska, J. Mol. Struct., 2002, 614, 227–232 CrossRef CAS ;
(b) S. M. T. Shaikh, J. Seetharamappa, S. Ashoka and P. B. Kandagal, Dyes Pigm., 2007, 73, 211–216 CrossRef CAS .
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC .
- M. Mitra, P. Raghavaiah and R. Ghosh, New J. Chem., 2015, 39, 200–205 RSC .
- W. S. Pierpoint, Biochem. J., 1969, 112, 609–616 CrossRef CAS PubMed .
-
(a) V. Selvarani, B. Annaraj, M. A. Neelakantan, S. Sundaramoorthy and D. Velmurugan, Spectrochim. Acta, Part A, 2012, 91, 329–337 CrossRef CAS PubMed ;
(b) B. Annaraj and M. A. Neelakantan, Eur. J. Med. Chem., 2015, 18, 1–8 CrossRef PubMed ;
(c) B. Annaraj, C. Balakrishnan and M. A. Neelakantan, J. Photochem. Photobiol., B, 2016, 160, 278–291 CrossRef CAS PubMed .
- A. Ohshima, A. Momotake and T. Arai, J. Photochem. Photobiol., A, 2004, 162, 473–479 CrossRef CAS .
- M. J. Gajewska, W. M. Ching, Y. S. Wen and C. H. Hung, Dalton Trans., 2014, 43, 14726–14736 RSC .
- L. K. Das, S. W. Park, S. J. Cho and A. Ghosh, Dalton Trans., 2012, 41, 11009–11017 RSC .
- R. L. Dutta and A. Syamal, Elements of Magnetochemistry, Affliated East-West Press, New Delhi, India, 2nd edn, 1993 Search PubMed .
-
(a) M. Barwiolek, E. Szlyk, T. M. Muziol and T. Lis, Dalton Trans., 2011, 40, 11012–11022 RSC ;
(b) M. Barwiolek, E. Szlyk, A. Surdykowski and A. Wojtczak, Dalton Trans., 2013, 42, 11476–11487 RSC .
-
(a) N. S. Gwaram, H. Khaledi and H. M. Ali, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m813 CAS ;
(b) V. Selvarani, B. Annaraj, M. A. Neelakantan, S. Sundaramoorthy and D. Velmurugan, Polyhedron, 2013, 54, 74–83 CrossRef CAS .
- D. Senthil Raja, N. S. P. Bhuvanesh and K. Natarajan, Inorg. Chem., 2011, 50, 12852–12866 CrossRef PubMed .
- V. M. Leovac, M. V. Rodic, L. S. Jovanovic, M. D. Joksovic, T. Stanojkovic, M. Vujcic, D. Sladic, V. Markovic and L. S. V. Jesic, Eur. J. Inorg. Chem., 2015, 2015, 882–895 CrossRef CAS .
-
(a) M. J. Waring, J. Mol. Biol., 1965, 13, 269–282 CrossRef CAS PubMed ;
(b) M. Zampakou, S. Balala, F. Perdih, S. Kalogiannis, I. Turel and G. Psomas, RSC Adv., 2015, 5, 11861–11872 RSC ;
(c) V. T. Yilmaz, C. Icsel, F. Suyunova, M. Aygun, N. Aztopal and E. Ulukaya, Dalton Trans., 2016, 45, 10466–10479 RSC .
-
(a) F. J. M. Almesa and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef ;
(b) B. D. Wang, Z. Y. Yang, Q. Wang, T. K. Cai and P. Crewdson, Bioorg. Med. Chem., 2006, 14, 1880–1888 CrossRef CAS PubMed ;
(c) S. Kathiresan, S. Mugesh, M. Murugan, F. Ahamed and J. Annaraj, RSC Adv., 2016, 6, 1810–1825 RSC .
-
(a) N. Chitrapriya, S. T. Kamatchi, M. Zeller, H. Lee and K. Natarajan, Spectrochim. Acta, Part A, 2011, 81, 128–134 CrossRef CAS PubMed ;
(b) J. R. Lakowicz and G. Webber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS PubMed ;
(c) B. C. Baguley and M. Le Bret, Biochemistry, 1984, 23, 937–943 CrossRef CAS PubMed .
- J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 2081–2088 CrossRef CAS .
- Y. B. Zeng, N. Yang, W. S. Liu and N. Tang, J. Inorg. Biochem., 2003, 97, 258–264 CrossRef CAS PubMed .
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed .
- V. Ramamurthy and K. S. Schanze, Organic and Inorganic Photochemistry, Marcel Dekker, Inc., 270 Madison Avenue, New York, 1998, vol. 2 Search PubMed .
- D. Xiao, L. Zhang, Q. Wang, X. Lin, J. Sun and H. Li, J. Lumin., 2014, 146, 218–225 CrossRef CAS .
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1990 Search PubMed .
-
(a) B. C. Baguley and M. L. Bret, Biochemistry, 1984, 23, 937–943 CrossRef CAS PubMed ;
(b) R. F. Pasternack, M. Caccam, B. Keogh, T. A. Stephenson, A. P. Williams and E. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS ;
(c) T. J. Meade, Met. Ions Biol. Syst., 1996, 32, 453–478 CAS .
-
(a) A. G. Zhang, Y. Z. Zhang, Z. M. Duan, K. Z. Wang, H. B. Wei, Z. Q. Bian and C. H. Huang, Inorg. Chem., 2011, 50, 6425–6436 CrossRef CAS PubMed ;
(b) A. Rajendran, C. J. Magesh and P. T. Perumal, Biochim. Biophys. Acta, 2008, 1780, 282–288 CrossRef CAS PubMed .
- Z. Bin, L. Yan, T. Xiaoning, X. Yinhua and X. Gang, J. Rare Earths, 2010, 28, 442–445 CrossRef .
-
(a) Y. Y. Lu, L. H. Gao, M. J. Han and K. Z. Wang, Eur. J. Inorg. Chem., 2006, 2006, 430–436 CrossRef ;
(b) S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1993, 32, 2573–2584 CrossRef CAS PubMed .
-
(a) A. Ghosh, P. Das, M. R. Gill, P. Kar, M. G. Walker, J. A. Thomas and A. Das, Chem.–Eur. J., 2011, 17, 2089–2098 CrossRef CAS PubMed ;
(b) C. V. Kumar and E. H. Asuncion, J. Am. Chem. Soc., 1993, 115, 8547–8553 CrossRef CAS ;
(c) P. Jaividhya, M. Ganeshpandian, R. Dhivya, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2015, 44, 11997–12010 RSC .
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2011, 46, 4584–4594 CrossRef CAS PubMed .
- D. S. Raja, G. Paramaguru, N. S. P. Bhuvanesh, J. H. Reibenspies, R. Renganathan and K. Natarajan, Dalton Trans., 2011, 40, 4548–4559 RSC .
- W. R. Ware, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS .
-
(a) J. R. Lakowicz, Fluorescence Quenching: Theory and Applications. Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999, pp. 53–127 Search PubMed ;
(b) X. Z. Feng, Z. Zhang, L. J. Yang, C. Wang and C. L. Bai, Talanta, 1998, 47, 1223–1229 CrossRef CAS PubMed .
- G. Z. Chen, X. Z. Huang, Z. Z. Zheng, J. G. Xu, and Z. B. Wang, Analysis Method of Fluorescence, Science Press, Beijing, 3rd edn, 1990 Search PubMed .
- J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 203–208 CAS .
- J. H. Tang, F. Luan and X. G. Chen, Bioorg. Med. Chem., 2006, 14, 3210–3217 CrossRef CAS PubMed .
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376–3387 CrossRef CAS PubMed .
-
(a) J. K. Barton, A. T. Danishefsky and J. M. Goldberg, J. Am. Chem. Soc., 1984, 106, 2172–2176 CrossRef CAS ;
(b) D. S. Sigman, Acc. Chem. Res., 1986, 19, 180–186 CrossRef CAS .
-
(a) S. Poornima, K. Gunasekaran and M. Kandaswamy, Dalton Trans., 2015, 44, 16361–16371 RSC ;
(b) S. Anbu, S. Shanmugaraju and M. Kandaswamy, RSC Adv., 2012, 2, 5349–5357 RSC .
- J. M. Kumar, M. M. Idris, G. Srinivas, P. K. Vinay, V. Meghah, M. Kavitha, C. R. Reddy, P. S. Mainkar, B. Pal, S. Chandrasekar and N. Nagesh, PLoS One, 2013, 8, e70798 Search PubMed .
- K. I. Ansari, S. Kasiri, J. D. Grant and S. S. Mandal, Dalton Trans., 2009, 8525–8531 RSC .
- C. Alapetite, T. Wachter, E. Sage and E. Moustacchi, Int. J. Radiat. Biol., 1996, 69, 359–369 CrossRef CAS PubMed .
- S. Tsuruya, S. I. Yanai and M. Masai, Inorg. Chem., 1986, 25, 141–146 CrossRef CAS .
- P. H. Rieger, in Electron Spin Resonance: Analysis and Interpretation, Royal Society of Chemistry, Cambridge, U.K., 2007 Search PubMed .
-
(a) A. Biswas, L. K. Das, M. G. B. Drew, G. Aromi, P. Gamez and A. Ghosh, Inorg. Chem., 2012, 51, 7993–8001 CrossRef CAS PubMed ;
(b) A. Guha, T. Chattopadhyay, N. D. Paul, M. Mukherjee, S. Goswami, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2012, 51, 8750–8759 CrossRef CAS PubMed ;
(c) L. Benisvy, R. Wanke, M. F. C. G. D. Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2011, 2011, 2791–2796 CrossRef CAS .
-
(a) T. Ghosh, J. Adhikary, P. Chakraborty, P. K. Sukul, M. S. Jana, T. K. Mondal, E. Zangrando and D. Das, Dalton Trans., 2014, 43, 841–852 RSC ;
(b) I. Majumder, P. Chakraborty, S. Das, H. Kara, S. K. Chattopadhyay, E. Zangrandod and D. Das, RSC Adv., 2015, 5, 51290–51301 RSC .
-
(a) A. I. Vogel, Textbook of quantitative inorganic analysis, Longmans, Green and Co. Ltd., London, 3rd edn, 1961, p. 366 Search PubMed ;
(b) A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki and E. Schwingel, Inorg. Chem., 2002, 41, 1788–1794 CrossRef CAS PubMed ;
(c) E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553–562 CrossRef CAS PubMed .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. J. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E. 01, Gaussian Inc., Wallingford, CT, 2004 Search PubMed .
- APEX2 “Program for Data Collection on Area Detectors”, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711–5373, USA Search PubMed .
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: The experimental part on binding interactions and catecholase activity are given. CCDC 1432869 and 1432870. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra20650f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a













