
Catalytic performance of a pyrolyzed graphene supported Fe–N–C composite and its application for acid direct methanol fuel cells†
Jingjing Xia,
Fang Wangb,
Riguo Meib,
Zhijie Gonga,
Xianping Fana,
Hui Yanga,
Liang Anc,
Qixing Wu *b and
Zhongkuan Luo*ab
*b and
Zhongkuan Luo*ab
aZhejiang California International NanoSystems Institute, Department of Materials Science and Engineering, Zhejiang University, 38 Zhe Da Road, Hangzhou, 310000, China
bShenzhen Key Laboratory of New Lithium-ion Batteries and Mesoporous Materials, College of Chemistry and Environmental Engineering, Shenzhen University, 3688 Nanhai Avenue, Nanshan District, Shenzhen, 518060, China. E-mail: qxwu@szu.edu.cn; lzk@szu.edu.cn; Fax: +86-755-26536141; Tel: +86-755-26557249
cDepartment of Mechanical Engineering, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China
First published on 16th September 2016
Abstract
In this work, a graphene supported Fe–N–C composite catalyst, synthesized by pyrolysis of graphene oxide (GO), graphitic carbon nitride (g-C3N4), ferric chloride (FeCl3) and carbon black (Vulcan XC-72), was evaluated for oxygen reduction reaction (ORR) in acid media. The introduction of carbon black was to separate the graphene sheets to enhance the specific surface area and thus improve the catalytic activity of the catalyst. The experimental results showed that the composite catalyst could yield an average electron transfer number of 3.85 and its onset and half-wave potentials for acidic ORR were only 56 and 69 mV smaller than those of Pt/C (40 wt% Pt) catalyst, respectively. The as-prepared catalyst was applied in an acid direct methanol fuel cell as the cathode catalyst and a peak power density of 11.72 mW cm−2 at 30 °C was demonstrated when feeding the anode and cathode with a 1 M methanol solution and air, respectively, suggesting its promising application.
1. Introduction
The direct methanol fuel cell (DMFC) has attracted significant attention and has been proposed as a future portable and mobile energy conversion device for its high energy density, easy handling and quiet operation as well as modular and scalable cell design.1–3 Presently, platinum and its alloys are widely used as oxygen reduction reaction (ORR) catalysts because of their ultra-high ORR activity.4,5 However, high cost and relatively poor durability prevent a large-scale commercialization of Pt-based catalysts for fuel cells.6–9 Therefore, replacing Pt-based catalysts by low-cost transition metals, including iron, manganese and cobalt, has been extensively investigated.10–12 In recent years, breakthroughs in enhancing the activities of such non-precious metal catalysts for ORR in alkaline media have been made and their catalytic performances have approached the level of Pt-based catalysts.13–17 In contrast, the activity of non-precious metal catalysts in acid media seems less pronounced than that in alkaline media and hence further improvements are needed.18,19 One promising candidate for promoting the ORR in acid media is Fe–N–C catalyst, which mainly contains non-pyrolyzed or pyrolyzed Fe-based macrocycle compound as well as pyrolyzed Fe/N-containing precursor materials.20–22 A large amount of literature confirms that catalysts, synthesized by pyrolyzing carbon and iron precursors with N-containing compounds, exhibit excellent ORR performance and catalyst stability.15,23–25 Further evidences26–28 indicate that Fe–N center and specific surface area are key factors that influence the ORR activity. Therefore, particular attentions are focused on synthesis of Fe–N–C catalyst with various iron precursors, N-sources and support materials with large specific surface areas. Jaouen et al.29 increased the N content of Fe–N–C catalysts through pyrolysis under NH3 atmosphere and found that an increase in the N content could be directly linked to improved ORR.Structurally analogous to graphite, graphitic carbon nitride (g-C3N4) exhibits a 2D sheet-like structure and it may be formed by triazines (C3N3) or heptazine (C6N7) interconnected via tertiary amines groups.30–32 It was firstly demonstrated by Lyth et al.33 that the pristine g-C3N4 could yield a much higher ORR activity than did carbon black in acidic electrolyte. In addition, the monolayer g-C3N4 is a competitive candidate of nitrogen source for Fe–N–C catalysts due to its large specific surface area (2500 cm2 g−1) and high nitrogen content (60.9 wt%). Meanwhile, g-C3N4 contains alleged “nitrogen pots” with six nitrogen lone-pair electrons, which are beneficial for metal inclusion.34 By introducing graphene into Fe–N–C catalyst, Byon et al.35 synthesized a novel Fe–N–C catalyst which delivered an excellent activity toward ORR in acid electrolyte. Wang et al.34 reported a new g-C3N4 based Fe–N–C catalyst by in situ polymerizing carbon black (Ketjenblack EC 600JD) supported Fe-doped g-C3N4 and such a catalyst displayed a decent performance in terms of activity and stability.
In this work, graphene supported Fe-g-C3N4 composites (Fe–N/C/rGO) separated by carbon black (Vulcan XC-72) were synthesized through chemical mixing and heat treatment by using reduced graphene oxide (rGO), Fe salt, g-C3N4 and carbon black as precursors, as shown in Fig. 1. The features of this composite catalyst includes: (a) facilitating the incorporation of Fe ions into g-C3N4 due to the interactions between cations and negatively charged nitrogen atoms;36 (b) tight interaction between graphene and g-C3N4 nanosheets due to their conjugated π-electron systems and similar aromatic structures; (c) alleviating the aggregation of nanosheets by an introduction of carbon black. The synthesized catalysts were evaluated experimentally in terms of its physical, chemical and electrochemical characteristics and the influences of pyrolysis temperatures and Fe contents were also discussed.
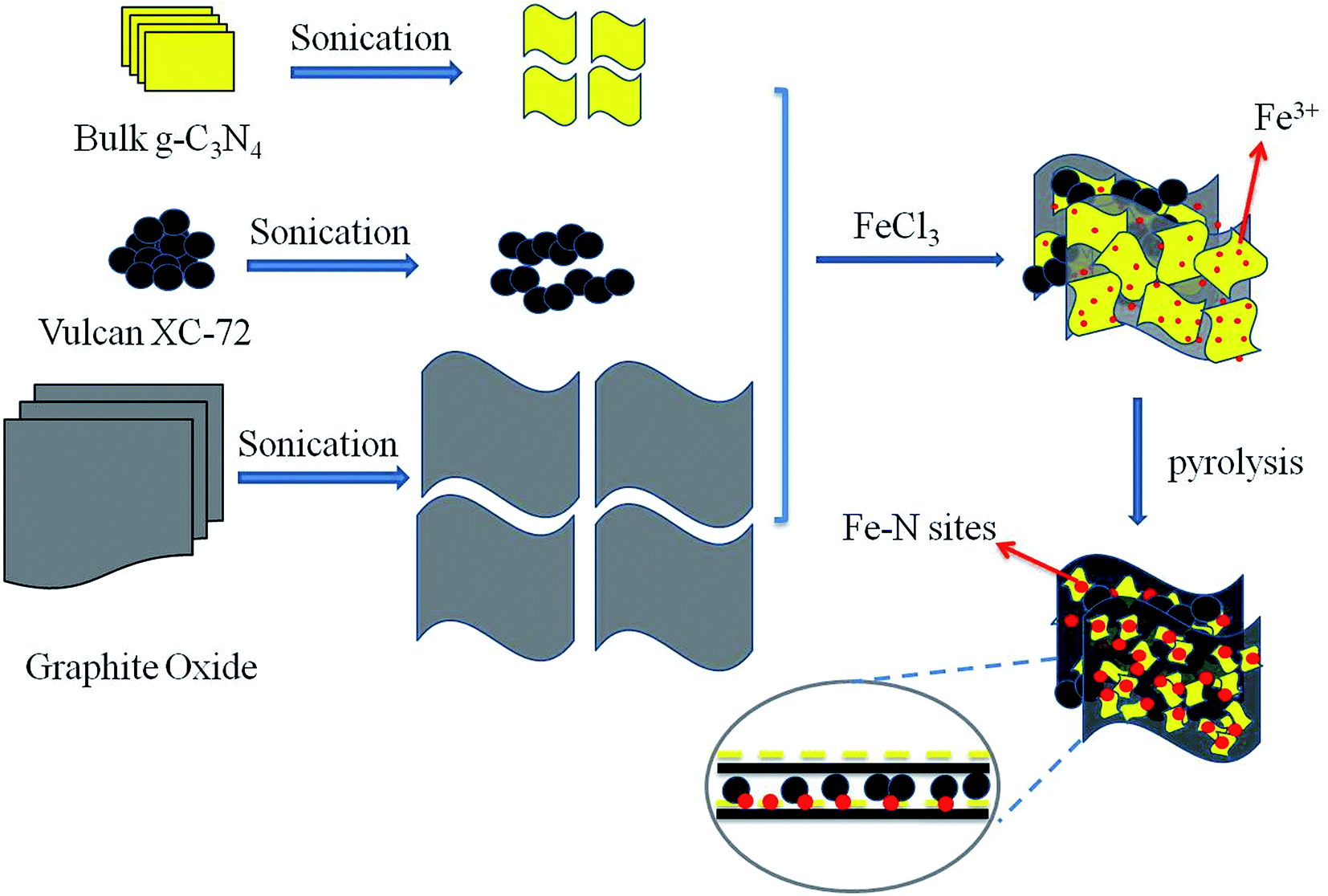 | ||
| Fig. 1 Schematic illustration for the preparation process of Fe–N/C/rGO catalysts. | ||
2. Experimental
2.1 Synthesis of graphene oxide (GO)
The GO was prepared by the modified Hummers method37 and it was briefly described as follows. 3 g graphite power (99.8 wt%, Aladdin), 360 mL 98% H2SO4, 40 mL H3PO4 (85 wt%, Aladdin) were mixed together through magnetic stirring, during which 18 g KMnO4 was added gradually. Then the mixture was kept at 50 °C with mild stirring for 24 hours. Subsequently, 400 mL ice composed of 1 wt% H2O2 (30 wt%, Aladdin) and 99 wt% H2O was added to the slurry to control its temperature at about 60 °C and stirring is kept until the temperature dropped to room temperature. To get rid of the impurity ions, the resulting product was washed and centrifugated several times. Finally, the obtained brown yellow graphite oxide was dispersed in water to form 1 mg mL−1 GO solution by ultrasonication.2.2 Synthesis of g-C3N4
The g-C3N4 was synthesized through thermal polymerization of melamine (99 wt%, Aladdin) and cyanuric acid (98 wt%, Aladdin)38 and the detailed procedure was described as follows. 4.2 g melamine and 8.6 g cyanuric acid were dispersed in 300 mL and 500 mL water respectively at 100 °C until the solutions became transparent. The melamine solution was then added slowly into the cyanuric acid solution and the mixture became milk white immediately. The mixture was stirred until the temperature dropped to room temperature and the obtained g-C3N4 precursor was put in quartz tube under argon atmosphere. The temperature of the furnace was elevated to 400 °C for 2 h with a heating rate of 10 °C min−1 and then rose to 550 °C for 3 h with a heating rate of 20 °C min−1. After cooling, the light yellow g-C3N4 was obtained.2.3 Synthesis of Fe–N/C/rGO catalyst
The Fe–N/C/rGO composite was prepared by the pyrolysis of carbon black, GO, g-C3N4 and FeCl3 (99.9 wt% Aladdin) mixture and the detailed procedure was summarized as follows. 0.05 g carbon black was dispersed in 10 mL N,N-dimethylformamide (DMF, Aladdin). 0.4 g g-C3N4 were dispersed in 200 mL water. Then, the g-C3N4 solution was added into 100 mL GO solution under stirring. After a few minutes, carbon powder solution was also added into the GO solution. Subsequently, 1 M HCl solution was added drop by drop to adjust the pH to 1–2. Finally, different weights of FeCl3 (Fe contents of 5, 10, 15 and 20 wt%) were added. The temperature of the solution was kept at 90 °C to evaporate the solvent to get the catalyst precursor. The obtained precursor was put in quartz tube and heated to 400 °C with a heating rate of 10 °C min−1 and stabilization of 2 h in argon atmosphere. Subsequently, the heating temperature was rose to various temperatures of 650, 750, 850 and 950 °C for 0.5 h with a heating rate of 20 °C min−1 to study the influence of pyrolysis temperature. After cooling, the black Fe–N/C/rGO was attained. For comparison, the catalyst without carbon black (Fe–N/rGO) or rGO (Fe–N/C) were also synthesized through the similar process with the identical g-C3N4 (0.4 g) and Fe (10 wt%) contents as well as pyrolysis temperature of 750 °C.In order to remove the unstable phases, the Fe–N/C/rGO catalyst with 10 wt% Fe and pyrolyzing at 750 °C was immersed in 0.5 M H2SO4 solution for 8 h. After being washed and dried, the leached Fe–N/C/rGO underwent a second pyrolysis by the same heating procedure. The catalyst with a second pyrolysis was labeled as Fe–N/C/rGO-LH.
2.4 Materials characterizations
Field emission scanning electron microscopy (FESEM, Hitachi SU-70) and transmission electron microscopy (TEM, Tecnai G2 Spirit 120 kV) were used to observe the morphologies of the prepared catalysts. X-ray diffraction (XRD) tests were performed by an apparatus (D8 Advance, Bruker) using Cu Kα radiation (λ = 0.15406 nm) to analyze the structure and composition. The specific surface areas of catalysts were measured by a gas sorption instrument (V-Sorb 2800TP, BELL). X-ray photoelectron spectroscopy (XPS) was performed on an instrument (PHI5300, PE) with Mg X-ray source (operating at 250 W, 14 kV). The contents of carbon, nitrogen, hydrogen were determined by an elemental analyzer (Vario EL, Elementar).2.5 Preparation of working electrodes and membrane electrode assemblies (MEA)
12 mg catalyst, 1 mL ethanol, 0.9 mL Milli-Q deionic water and 0.1 mL nafion solution (5 wt%) were mixed together and the mixture was sonicated by a ultrasonic processor (Sonics VCX 750) for 30 min to prepare a homogenous catalyst ink with a concentration of 6 mg mL−1. Then 10 μL ink was coated onto the glassy carbon disk electrode and the resulting loading of the catalyst was 0.8 mg cm−2. Commercial Pt/C catalysts (40 wt% Pt) with a loading of 0.4 mg cm−2 were also tested for comparison.To prepare the cathode of the MEA, the catalyst ink was sprayed on the gas diffusion layer (GDL) by an automatic ultrasonic spraying machine (Siansonic). The catalyst loading for the cathode was 5.0 mg cm−2 and the Nafion content was 50 wt%. The anode electrode was the commercial PtRu electrode (Alfa Aesar 45374) with a metal loading of 4 mg cm−2. To form the MEA, the cathode and anode, with the active areas of 5 cm2, was sandwiched between a Nafion 212 membrane under 3.2 MPa at 140 °C for 3 min.
2.6 Electrochemical tests
All the ORR tests were carried out by a CHI 760d electrochemical station and RRDE-3A rotating disk electrode under room temperature. Linear scanning voltammetry (LSV) was performed with a standard three-electrode electrochemical cell. The glassy carbon electrode, Pt wire and Ag/AgCl electrode (saturated KCl solution) were used as the working electrode, counter electrode and reference electrode, respectively. A scan rate of 5 mV s−1 and a rotation rate of 1600 rpm were used during LSV tests. 0.5 M H2SO4 solution was used for the electrolyte and it was pre-purged and saturated with high purity O2. The measured ORR currents of various catalysts in O2-saturated electrolyte were corrected by subtracting their respective background currents in N2-saturated electrolyte. All potentials in this work were corrected by ERHE = EAg/AgCl + EθAg/AgCl + 0.059pH39 and given versus reversible hydrogen electrode (RHE).The DMFC was assembled with two gold-plated current collectors, two grooved graphite blocks (Poco graphite) with serpentine flow fields and a MEA. The fuel cell test was performed by an Arbin BT-5HC testing system.40–42 During polarization test, the anode and cathode were fed by 2.5 mL min−1 methanol solution and 100 sccm compressed air, respectively. To attain stable polarization curves, the DMFCs were discharged at a series of predefined current until reproducible data was achieved. The operation temperature of the cell was kept at 30 °C.
3. Results and discussion
3.1 Physical and chemical characterizations
Fig. 2(a–e) show the morphologies of Fe–N/C, Fe–N/rGO, Fe–N/C/rGO and Fe–N/C/rGO-LH. It is seen that the Fe–N/C catalyst is composed of aggregated particles and several small Fe–N sheets derived from pyrolysis of FeCl3 and g-C3N4. The Fe–N/rGO presented in Fig. 2(b) shows a stacked and wrinkled structure resulting from the aggregation of 2D rGO and g-C3N4. For the Fe–N/C/rGO in Fig. 2(c), it is seen that the large rGO sheets are separated by carbon particles, which is beneficial for forming loose structures and enhancing the surface area. In addition, it is interesting to observe that there is no substantial change in the morphology between Fe–N/C/rGO and Fe–N/C/rGO-LH, suggesting acid leaching and second pyrolysis have little effect on the macroscopic morphology. However, it is worth mentioning that the morphology of Fe–N/C/rGO catalyst is significantly affected by the Fe content.43 As shown in Fig. S1,† when increasing the Fe content, the agglomeration in Fe–N/C/rGO catalysts becomes more serious due to the formation of excessive Fe-containing composites.43 In contrast to the effect of Fe content, the influence of the pyrolysis temperature on the morphology is negligible as shown in Fig. S2.† The Fe–N/C/rGO-LH is further identified by TEM in Fig. 2(e). It can be seen that Fe–N sheets are adhered to the rGO sheets due to the π–π stacking effect and the carbon particles are distributed dispersely among the rGO sheets. | ||
| Fig. 2 FESEM images of Fe–N/C (a), Fe–N/rGO (b), Fe–N/C/rGO (c) and Fe–N/C/rGO-LH (d); TEM images of Fe–N/C/rGO-LH (e). | ||
As shown in Fig. 3(a), all the N2 adsorption/desorption isotherms of the prepared catalysts show remarkable hysteresis loops of IV isotherm according to IUPAC's classification, indicating the mesoporous structures of the catalysts.44 From the pore size distributions shown in Fig. 3(b), one can see that the pore diameters of all catalysts centered mainly at about 4–5 nm. The specific surface areas and pore volumes of prepared catalysts are listed in Table 1. It is found that Fe–N/rGO exhibits the lowest specific surface area and pore volume (117 m2 g−1 and 0.36 cm3 g−1, respectively), likely resulting from the aggregation of the rGO sheets. In contrast, such values of Fe–N/C/rGO are increased by more than 2 times (256 m2 g−1 and 0.87 cm3 g−1, respectively), suggesting the introduction of carbon particles could alleviate the aggregation of rGO sheets. It is important to note that the surface area could be influenced by the Fe content. As evident in Table S1,† although the surface areas of Fe–N/C/rGO catalysts remain nearly unchanged when increasing the content of Fe from 5 to 10 wt%, a further increase of the Fe content from 10 to 20 wt% results in significant decreases in both surface areas and pore volumes as excessive Fe-containing composites may be formed.43 This result suggests that the amount of g-C3N4 in Fe–N/C/rGO catalysts can only afford coordination sites for about 10 wt% Fe. Moreover, it is found that the pyrolysis temperature also affects the surface area and pore volume of the catalysts. As shown in Table S1,† an increased pyrolysis temperature leads to an improved SBET and Vpore in the temperature range of 650–750 °C, but further increasing the temperature from 750 to 950 °C results in a gradual decrease in SBET and Vpore, possibly because the gaseous decomposition products during pyrolysis somehow destroy the mesoporous structure. Based on the above N2 adsorption/desorption results, the optimal content of Fe and heating temperature used in preparation of the in-house Fe–N/C/rGO catalyst are 10 wt% and 750 °C, respectively. To further increase SBET and Vpore, the Fe–N/C/rGO catalyst is treated with H2SO4 and second pyrolysis45 to remove the unstable phases presented on the catalyst surface. As shown in Table 1, its SBET and Vpore are further increased to 343 m2 g−1 and 1.00 cm3 g−1, respectively. The large SBET and Vpore of Fe–N/C/rGO-LH are beneficial for promoting the exposure of catalytic areas toward ORR.13,46
 | ||
| Fig. 3 N2 adsorption/desorption isotherms (a) and BJH pore size distributions (b) of Fe–N/C, Fe–N/rGO, Fe–N/C/rGO and Fe–N/C/rGO-LH. | ||
| Samples | SBET (m2 g−1) | Vpore (cm3 g−1) |
|---|---|---|
| Fe–N/C | 213 | 0.48 |
| Fe–N/rGO | 117 | 0.36 |
| Fe–N/C/rGO | 256 | 0.87 |
| Fe–N/C/rGO-LH | 343 | 1.00 |
XRD patterns of all the catalysts are shown in Fig. 4(a). The characteristic diffraction peaks for Fe–N/C, Fe–N/rGO and Fe–N/C/rGO can be confirmed as different Fe-containing crystalline phases such as Fe3C, Fe, Fe3N and Fe4N. In addition, it is found that Fe3O4 is formed during the pyrolysis under argon atmosphere. According to the XRD patterns of catalyst precursors (Fig. S3†), all the precursors contain certain amount of Fe2O3·H2O, which is probably produced by the hydrolysis of FeCl3 during solvent evaporation. Hence, it is guessed that the observed Fe3O4 in the catalysts is resulted from the pyrolysis of Fe2O3·H2O. Fig. S4† shows the XRD patterns of Fe–N/C/rGO catalysts with different Fe contents and pyrolysis temperatures. When the Fe content increases from 5 wt% to 20 wt%, Fe3N and Fe4N gradually disappear and Fe3C is generated when the Fe content is higher than 10 wt%, indicating a competition between the formation of Fe–C and Fe–N composites.34 Furthermore, it is seen in Fig. S4(b)† that when the pyrolysis temperature is higher than 750 °C, Fe3N and Fe4N seem to transform to FeN0.056 due to the loss of nitrogen at higher temperatures.45 Another important phenomenon should be mentioned is that the unstable species on the catalyst are removed by acid washing and second pyrolysis: only Fe and FeN0.056 are remained in Fe–N/C/rGO-LH (Fig. 4(a)). This indicates that metallic Fe is probably covered by the carbon shell and FeN0.056 is likely resulted from the transformation of Fe3N and Fe4N during the second pyrolysis.47 As the Fe-containing species are unstable in the acid condition, which may cause the contamination of a proton exchanging membrane,45 leading to a poor life time of a fuel cell, thus, the acid and heat treatments are favorable for the practical application of fuel cells. The chemical compositions of the prepared catalysts are analyzed by XPS, which are displayed in Fig. 4(b). Clearly, only C1s, N1s, O1s and Fe2p regions are detected in all catalysts, suggesting that the impurity is negligible. It has been known that nitrogen content is an important factor for evaluating the Fe–N catalysts26,28 and hence elemental analysis is performed and the nitrogen contents are determined to be 2.14, 2.98, 5.8 and 4.45 wt%, respectively, for Fe–N/C, Fe–N/rGO, Fe–N/C/rGO and Fe–N/C/rGO-LH. As nitrogen content in the catalyst can indirectly reflect the Fe–N active sites, it is believed that Fe–N/C/rGO and Fe–N/C/rGO-LH may possess better ORR activity than do the Fe–N/C and Fe–N/rGO. To further analyze the chemical states of nitrogen in the catalysts, the high-resolution N1s spectra are displayed in Fig. 4(c–f). The fitted N1s plots can be divided into four nitrogen species, including oxidic N, graphitic N, pyrrolic N and pyridinic N at about 403.2, 401.1, 400.1 and 398.5 eV, respectively.48 It is reported that pyrrolic-N and pyridinic-N refer to nitrogen atoms at the edge of carbon planes, which can coordinate with iron to form Fe–N active sites for ORR.45,49,50 The quantitative analysis of different N species in various catalysts is summarized in Table 2. It can be found that the total contents of pyrrolic-N and pyridinic-N in Fe–N/C/rGO and Fe–N/C/rGO-LH are substantially large, which further suggests their high activities toward ORR.
 | ||
| Fig. 4 XRD patterns (a) and XPS survey (b) of as-prepared catalysts; high resolution N1s spectra of Fe–N/C (c), Fe–N/rGO (d), Fe–N/C/rGO (e), Fe–N/C/rGO-LH (f). | ||
| Samples | Pyridinic N (%) | Pyrrolic N (%) | Graphitic N (%) | Oxidic N (%) |
|---|---|---|---|---|
| Fe–N/C | 8.3 | 27.7 | 37.2 | 26.8 |
| Fe–N/rGO | 14.7 | 29.2 | 22.1 | 34.0 |
| Fe–N/C/rGO | 33.9 | 26.2 | 19.1 | 20.8 |
| Fe–N/C/rGO-LH | 34.4 | 23.9 | 23.5 | 18.2 |
3.2 ORR characterization
The results of LSV tests for various catalysts are shown in Fig. 5(a). Compared with Fe–N/C, the onset potentials of Fe–N/rGO and Fe–N/C/rGO shift positively, demonstrating that the rGO matrix may provide unique carbon chemistry to promote the formation of Fe–N groups.35 In addition, it is found that Fe–N/rGO exhibits a substantially lower limiting current density than does the Fe–N/C because of the low surface area in Fe–N/rGO. With the introduction of carbon particles into Fe–N/rGO, the surface area and pore volume can be drastically increased (Table 1) and thus the limiting current density of Fe–N/C/rGO is significantly improved as shown in Fig. 5(a), in good agreement with previous elemental analysis and XPS results. The effects of Fe content and pyrolysis temperature on ORR activity are displayed in Fig. S5.† Compared with 5 wt% Fe content, 10 wt% Fe in the Fe–N/C/rGO catalyst can yield a higher half-wave potential and limiting current density likely because more Fe–N active sites are formed with a higher Fe content. However when Fe content is further increased from 10 to 20 wt%, the half-wave potential and limiting current density decrease gradually owing to the decreases of N content and surface area. Additionally, it is seen in Fig. S5(b)† that the onset potential is considerably low when the pyrolysis temperature is 650 °C probably due to the incomplete decomposition of g-C3N4, whereas increasing the pyrolysis temperature from 750 to 950 °C leads to an gradual decrease of onset potential owing to losses of surface area and nitrogen as evident in Tables S1 and 2† Another finding in Fig. 5(a) is that Fe–N/C/rGO-LH exhibits a high onset and half-wave potentials (E1/2) of 0.860 V and 0.717 V toward ORR, respectively. Such potentials are only 56 mV and 69 mV smaller than those of commercially available Pt/C and are comparable to those of recent advanced Fe-based catalysts for acidic ORR (Table S3†). The enhanced activity of Fe–N/C/rGO-LH may be attributed to the encapsulated Fe species and second pyrolysis, which modify the carbon matrix and active sites.51 The intrinsic activity of the catalyst is obtained based on the Koutecky–Levich equation.34 The ORR mass activity of the prepared catalysts at 0.75 V are shown in Fig. 5(b). As expectedly, Fe–N/C/rGO-LH delivers a mass activity of 1.045 A g−1, much higher than those of other catalysts. To further evaluate the ORR kinetics, Tafel slops are shown in the inset of Fig. 5(b). Tafel slops for Fe–N/C, Fe–N/rGO, Fe–N/C/rGO and Fe–N/C/rGO-LH at potentials lower than 0.8 V are 63, 72, 69 and 63 mV dec−1, respectively, which are similar to the value of commercial Pt/C (60 mV dec−1) in low overpotential regions. These results indicate the reaction rates of all catalysts is controlled by the first electron transfer process and the oxygen adsorption mechanism is Temkin adsorption isotherm in the measured potential range.45,52 | ||
| Fig. 5 LSV curves of Fe–N/C, Fe–N/rGO, Fe–N/C/rGO, Fe–N/C/rGO-LH and Pt/C in O2-saturated 0.5 M H2SO4 solution at the rotating speed of 1600 rpm and the scan rate of 5 mV s−1 (a); ORR mass activities of Fe–N/C, Fe–N/rGO, Fe–N/C/rGO and Fe–N/C/rGO-LH at 0.75 V vs. RHE. The inset is Tafel plot (b); LSV curves of Fe–N/C/rGO-LH with various rotation rates at the scan rate of 5 mV s−1. The inset shows the corresponding K–L plots at a potential range of 0.2 to 0.4 V vs. RHE (c); current–time chronoamperometric response of Fe–N/C/rGO-LH and Pt/C under the rotating speed of 1600 rpm in O2-saturated 0.5 M H2SO4 at 0.5 V vs. RHE (d). As-prepared catalyst loading: 0.8 mg cm−2; Pt/C loading: 0.4 mg cm−2. | ||
Fig. 5(c) shows the LSV plots of Fe–N/C/rGO-LH under various rotation rates (400, 900, 1600 and 2500 rpm). The limiting current density increases from −1.73 to −5.02 mA cm−2 with an increase in the rotation rates. It is found that the peak currents in the LSV curves are observed at low rotation speeds, whereas no obvious peak current is seen at a high rotation speed of 2500 rpm. This phenomenon can be attributed to the possible solid phase diffusion and in-plane diffusion processes at a high catalyst loading.53 The inset of Fig. 5(c) displays the Koutecky–Levich plots for the Fe–N/C/rGO-LH catalyst with various potentials from 0.2 to 0.4 V. The electron transfer number during ORR, n, is estimated by the Koutecky–Levich equation:
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 486C mol−1), CO2 the bulk concentration of oxygen (1.4 × 10−6 mol mL−1), DO2 the diffusivity of oxygen in 0.5 M H2SO4 (1.15 × 10−5 cm2 s−1) and v the kinematic viscosity of the electrolyte (1.07 × 10−2 cm2 s−1).54,55 According to the eqn (1), the values of n for Fe–N/C/rGO-LH at 0.2, 0.25, 0.3, 0.35 and 0.4 V are 3.86, 3.88, 3.86, 3.81 and 3.83, respectively, implying that O2 is mainly reduced to H2O through a four-electron transfer pathway in acid media.54 The short-term stabilities of Fe–N/C/rGO-LH and commercial Pt/C catalysts in acid are evaluated and compared by the current–time chronoamperometric test at 0.5 V vs. RHE and 1600 rpm. The results in Fig. 5(d) show that the current density of Fe–N/C/rGO-LH maintains as high as 90.3%, while the current density of Pt/C only remains 68.8% after 20000 s, demonstrating a good stability of Fe–N/C/rGO-LH.
486C mol−1), CO2 the bulk concentration of oxygen (1.4 × 10−6 mol mL−1), DO2 the diffusivity of oxygen in 0.5 M H2SO4 (1.15 × 10−5 cm2 s−1) and v the kinematic viscosity of the electrolyte (1.07 × 10−2 cm2 s−1).54,55 According to the eqn (1), the values of n for Fe–N/C/rGO-LH at 0.2, 0.25, 0.3, 0.35 and 0.4 V are 3.86, 3.88, 3.86, 3.81 and 3.83, respectively, implying that O2 is mainly reduced to H2O through a four-electron transfer pathway in acid media.54 The short-term stabilities of Fe–N/C/rGO-LH and commercial Pt/C catalysts in acid are evaluated and compared by the current–time chronoamperometric test at 0.5 V vs. RHE and 1600 rpm. The results in Fig. 5(d) show that the current density of Fe–N/C/rGO-LH maintains as high as 90.3%, while the current density of Pt/C only remains 68.8% after 20000 s, demonstrating a good stability of Fe–N/C/rGO-LH.
3.3 DMFC performance
The morphology of the cathode prepared with Fe–N/C/rGO-LH catalysts is presented in Fig. 6(a). Interestingly, the coating of Fe–N/C/rGO-LH catalysts on the GDL is quite uniform without any visible macro-cracks. This may be beneficial for reducing the water flooding in the catalyst layer as water tends to accumulate in the large cracks (or pores). In the inset of Fig. 6(a), the Fe–N/C/rGO-LH cathode exhibits a loose network structure constructed by a number of sheets, which might facilitate the transport of oxygen.13 The performances of the DMFC using Fe–N/C/rGO-LH as cathode catalysts with different methanol concentrations are shown in Fig. 6(b). It is seen that the peak power densities are 11.72, 9.58 and 9.28 mW cm−2, respectively, at the concentrations of 1, 2 and 4 M. Such power densities are roughly the half of those with commercial Pt/C catalysts under similar operating conditions, implying a promising application of the catalyst in a practical fuel cell; the peak power density of a liquid-feed DMFC with air/oxygen as oxidant and Pt/C as cathode catalysts generally falls in the range of 15–25 mW cm−2 at 30 °C.56–59 In addition, it is found in Fig. 6(b) that when the methanol concentration increases from 2 M to 4 M, the polarization curves are almost overlapped at small current densities (<50 mA cm−2) where methanol crossover dominates. Hence, as compared with Pt/C which may be readily poisoned by the permeated methanol from the anode and thus creates a mixed potential, the Fe–N/C/rGO-LH cathode shows a better tolerance against methanol poisoning, further demonstrating its desirable application in DMFCs. | ||
| Fig. 6 Morphologies of Fe–N/C/rGO-LH cathode, the inset is the enlarged view (a); polarization and power density plots for direct methanol fuel cells with Fe–N/C/rGO-LH as cathode catalysts at 30 °C (b). MEA active area: 5 cm2; Nafion 212 membrane; cathode catalyst loading: 5 mg cm2; anode catalyst: PtRu with 4 mg cm−2. | ||
4. Conclusion
A composite Fe–N–C catalyst is synthesized through chemical mixing and heat treatment by using rGO, Fe salt, g-C3N4 and carbon black as precursors. The introduction of carbon particles impedes the stacks of GO and g-C3N4 to remarkably increase the surface area and pore volume. After acid treatment and second pyrolysis, the catalyst yields an excellent catalytic activity with the onset and half-wave potentials of 56 and 69 mV smaller than those of Pt/C and guarantees an approximately four-electron transfer pathway during ORR. The peak power density of the DMFC with Fe–N/C/rGO-LH as the cathode catalyst can reach 11.72 mW cm−2 at 30 °C, indicating its promising application in practical DMFCs.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51306125), Shenzhen Science and Technology Fund (JCYJ20140828163634002, KQCX20140519105122378 and JCYJ20150324141711693) and Natural Science Foundation of SZU (No. 827-000015).References
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS.
- E. Negro, A. H. A. M. Videla, V. Baglio, A. S. Arico, S. Specchia and G. J. M. Koper, Appl. Catal., B, 2015, 166, 75–83 CrossRef.
- H. Tang, H. Yin, J. Wang, N. Yang, D. Wang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 5585–5589 CrossRef CAS PubMed.
- R. Othman, A. L. Dicks and Z. H. Zhu, Int. J. Hydrogen Energy, 2012, 37, 357–372 CrossRef CAS.
- N. Jung, D. Y. Chung, J. Ryu, S. J. Yoo and Y. E. Sung, Nano Today, 2014, 9, 433–456 CrossRef CAS.
- B. P. Vinayan and S. Ramaprabhu, Nanoscale, 2013, 5, 5109–5118 RSC.
- N. Karthikeyan, B. P. Vinayan, M. Rajesh, K. Balaji, A. K. Subramani and S. Ramaprabhu, Fuel Cells, 2015, 15, 278–287 CrossRef CAS.
- H. M. Wu, D. Wexler and H. K. Liu, Mater. Chem. Phys., 2012, 136, 845–849 CrossRef CAS.
- D. C. Higgins and Z. W. Chen, Can. J. Chem. Eng., 2013, 91, 1881–1895 CrossRef CAS.
- J. H. Lee, M. J. Park, S. J. Yoo, J. H. Jang, H. J. Kim, S. W. Nam, C. W. Yoon and J. Y. Kim, Nanoscale, 2015, 7, 10334–10339 RSC.
- Q. Liu and J. Y. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS PubMed.
- C. Domínguez, F. Pérez-Alonso, M. A. Salam, J. L. Gómez-de-la-Fuente, S. Abdel-Thabaiti, S. Basahel, M. Peña, J. Fierro and S. Rojas, Int. J. Hydrogen Energy, 2014, 39, 5309–5318 CrossRef.
- Y. C. Wang, Y. J. Lai, L. Song, Z. Y. Zhou, J. G. Liu, Q. Wang, X. D. Yang, C. Chen, W. Shi and Y. P. Zheng, Angew. Chem., Int. Ed., 2015, 127, 10045–10048 CrossRef.
- L. Yang, N. Larouche, R. Chenitz, G. Zhang, M. Lefèvre and J. P. Dodelet, Electrochim. Acta, 2015, 159, 184–197 CrossRef CAS.
- J. Tian, A. Morozan, M. T. Sougrati, M. Lefèvre, R. Chenitz, J. P. Dodelet, D. Jones and F. Jaouen, Angew. Chem., Int. Ed., 2013, 125, 7005–7008 CrossRef.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- S. Yuan, J. L. Shui, L. Grabstanowicz, C. Chen, S. Commet, B. Reprogle, T. Xu, L. Yu and D. J. Liu, Angew. Chem., Int. Ed., 2013, 125, 8507–8511 CrossRef.
- X. Li, G. Liu and B. N. Popov, J. Power Sources, 2010, 195, 6373–6378 CrossRef CAS.
- C. W. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- Z. Xiang, Y. Xue, D. Cao, L. Huang, J. F. Chen and L. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433–2437 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J. P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- X. Chen, F. Li, N. L. Zhang, L. An and D. G. Xia, Phys. Chem. Chem. Phys., 2013, 15, 19330–19336 RSC.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach and S. Mukerjee, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
- U. I. Kramm, M. Lefeèvre, N. Larouche, D. Schmeisser and J. P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978–985 CrossRef CAS PubMed.
- F. Jaouen and J. P. Dodelet, Electrochim. Acta, 2007, 52, 5975–5984 CrossRef CAS.
- C. Domínguez, F. Pérez-Alonso, M. A. Salam, J. Fuente, S. Al-Thabaiti, S. Basahel, M. Peña, J. Fierro and S. Rojas, Int. J. Hydrogen Energy, 2014, 39, 5309–5318 CrossRef.
- F. Charreteur, S. Ruggeri, F. Jaouen and J. P. Dodelet, Electrochim. Acta, 2008, 53, 6881–6889 CrossRef CAS.
- F. Jaouen, F. Charreteur and J. Dodelet, J. Electrochem. Soc., 2006, 153, A689–A698 CrossRef CAS.
- Y. H. Qi, L. Liu, Y. H. Liang, J. S. Hu and W. Q. Cui, Prog. Chem., 2015, 27, 38–46 Search PubMed.
- G. Algara-Siller, N. Severin, S. Y. Chong, T. Bjorkman, R. G. Palgrave, A. Laybourn, M. Antonietti, Y. Z. Khimyak, A. V. Krasheninnikov, J. P. Rabe, U. Kaiser, A. I. Cooper, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2014, 53, 7450–7455 CrossRef CAS PubMed.
- Y. T. Gong, M. M. Li and Y. Wang, ChemSusChem, 2015, 8, 931–946 CrossRef CAS PubMed.
- S. M. Lyth, Y. Nabae, S. Moriya, S. Kuroki, M. A. Kakimoto, J. I. Ozaki and S. Miyata, J. Phys. Chem. C, 2009, 113, 20148–20151 CAS.
- M. Q. Wang, W. H. Yang, H. H. Wang, C. Chen, Z. Y. Zhou and S. G. Sun, ACS Catal., 2014, 4, 3928–3936 CrossRef CAS.
- H. R. Byon, J. Suntivich and Y. Shao-Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef CAS.
- S. W. Cao, J. X. Low, J. G. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Y. Liao, S. Zhu, J. Ma, Z. Sun, C. Yin, C. Zhu, X. Lou and D. Zhang, ChemCatChem, 2014, 6, 3419–3425 CrossRef CAS.
- H. Yang, Y. Zhang, F. Hu and Q. Wang, Nano Lett., 2015, 15, 7616–7620 CrossRef CAS PubMed.
- Q. X. Wu, L. An, X. H. Yan and T. S. Zhao, Electrochim. Acta, 2014, 133, 8–15 CrossRef CAS.
- X. X. Zhao, W. X. Yuan, Q. X. Wu, H. Y. Sun, Z. K. Luo and H. D. Fu, J. Power Sources, 2015, 273, 517–521 CrossRef CAS.
- Q. X. Wu, H. Y. Li, W. X. Yuan, Z. K. Luo, F. Wang, H. Y. Sun, X. X. Zhao and H. D. Fu, Appl. Energy, 2015, 160, 146–152 CrossRef CAS.
- L. Yang, Y. Su, W. Li and X. Kan, J. Phys. Chem. C, 2015, 119, 11311–11319 CAS.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- F. Afsahi and S. Kaliaguine, J. Mater. Chem. A, 2014, 2, 12270–12279 CAS.
- Z. Y. Wu, X. X. Xu, B. C. Hu, H. W. Liang, Y. Lin, L. F. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
- U. I. Kramm, I. Herrmann-Geppert, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2011, 115, 23417–23427 CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- M. Chen, J. Liu, W. Zhou, J. Lin and Z. Shen, Sci. Rep., 2015, 5, 17396 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Appl. Catal., B, 2009, 93, 156–165 CrossRef CAS.
- Y. Wu, J. Zang, L. Dong, Y. Zhang and Y. Wang, J. Power Sources, 2016, 305, 64–71 CrossRef CAS.
- M. Zhou, C. Yang and K. Y. Chan, Adv. Energy Mater., 2014, 4, 1400840 CrossRef.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron and W. Schuhmann, J. Phys. Chem. C, 2009, 113, 14302–14310 CAS.
- K. Hsueh, E. Gonzalez and S. Srinivasan, Electrochim. Acta, 1983, 28, 691–697 CrossRef CAS.
- F. Xie, C. Chen, H. Meng and P. Shen, Fuel Cells, 2007, 7, 319–322 CrossRef CAS.
- X. Zhao, X. Fan, S. Wang, S. Yang, B. Yi, Q. Xin and G. Sun, Int. J. Hydrogen Energy, 2005, 30, 1003–1010 CrossRef CAS.
- H. Yang, T. Zhao and Q. Ye, J. Power Sources, 2005, 139, 79–90 CrossRef CAS.
- N. Nakagawa and Y. Xiu, J. Power Sources, 2003, 118, 248–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20593c |
| This journal is © The Royal Society of Chemistry 2016 |