
Supramolecular porous ionic network based on triazinonide and imidazolium: a template-free synthesis of meso-/macroporous organic materials via a one-pot reaction-assembly procedure†
Ze-Huan Heia,
Gan-Lin Songa,
Chen-Yu Zhaoa,
Wenhao Fanb and
Mu-Hua Huang*a
aSchool of Materials Science and Engineering, Beijing Institute of Technology, Beijing, 100081, China. E-mail: mhhuang@bit.edu.cn
bBeijingCenter for Physical & Chemical Analysis, Beijing, 100089, China
First published on 20th September 2016
Abstract
A supramolecular porous ionic network (SPIN-1) was designed and prepared using a one-pot procedure, involving the quaternization of triimidazole triazine with cyanuric chloride followed by hydrolysis and in situ assembly. The structure of SPIN-1 was characterized by FT-IR, solid-state 13C CP/TOSS NMR, elemental analysis and XPS. SPIN-1 shows a BET surface area up to 263 m2 g−1 (average pore size 11 nm), and a CO2 capture capacity of 24.7 cm3 g−1, which is competitive for Carbon Capture and Storage (CCS).
Introduction
Porous organic polymers (POPs) linked by a covalent bond have attracted much attention, since they show widespread applications in adsorption,1–5 catalysis,6–9 luminescence,10,11 batteries12,13 and so on.14,15 Among them, Ionic porous organic polymers16,17 combine the unique features of ionic compounds with the common properties of porous materials. Based on the charges involved in the backbone of porous polymers, the ionic porous organic polymers can be divided into cationic and anionic types. Cationic type porous organic materials usually contain positive charge in the backbone, such as ammonium,18 pyridinium,19 imidazolium,20 phosponium21 and so on. Based on the covalent triazine frameworks (CTF),22 the imidazolium CTF (Fig. 1) was developed by Dai's group20 for ion exchange. A pyridinium-based porous polymer named PAF-50 (Fig. 1) was designed and synthesized by the Zhu group.15 Correspondingly, Anionic type porous organic materials usually contain negative charge in the backbone, such as boronate. Tetraphenylborate network was made by Long group for single ion conductor, whose ionic conductivity reached 10−3 S cm−1.13 As far as synthetic methods are concerned, the charged backbone in the porous organic materials were are usually constructed through covalent bond formation, i.e., the polymerization on the charged monomer. In addition to covalent bond formation, a supramolecular interaction was introduced to construct the charged porous organic materials. For example, a novel supramolecular organic frameworks (SOFs) by self-assembly of cucurbit[8]uril and multi-armed pyridiniums were developed.23–25 | ||
| Fig. 1 Structure of some representative ionic porous organic materials, supramolecular ionic networks and SPIN-1. | ||
Actually, the ionic bonds as the reversible non-covalent links was first used by Grinstaff26,27 group to construct the supramolecular ionic networks (Fig. 1) by combining multicationic and multianionic compounds. And it was further developed by Mecerreyes et al.,28,29 though the porous property was not investigated in those work. Hierarchical organic porous materials30,31 have enhanced properties such as good pore volume, large accessible space and interconnected hierarchical porosity. By polymeric multications and multianions, a micro/mesoporous polymeric ionic liquid complex (PILC)32 (Fig. 1) can be prepared, possibly owing to the relatively less flexible polymer chain compared to small molecules. Mesoporous structures are especially desirable in heterocatalysis because they are favorable to the mass transfer of reactants.6 The synthesis of the mesoporous POPs usually requires templates such as SiO2 (ref. 33) and surfactants,34 which add troubles to remove templates. A template-free synthesis thus attracted attention recently. Hazo-POPs were reported by Liu group35 and divinylbenzene-based mesoporous POPs were developed by Xiao group36 without using template. Hence, we became interested in the template-free synthesis of porous organic polymers based on supramolecular ionic network.
We wonder if the further introduction of rigid element to the supramolecular ionic network will help the construction of porosity of the materials, e.g. an alternative anion and cation in the conjugated system plus π–π stacking.37 The trisubstituted triazine with imidazolium38 was reported to hydrolyze to give structures with some of their counter ion delocalized to form less charged conjugated structure.39 Thus, the structure of SPIN-1 (Fig. 1) came into our mind. In this paper, we would like to present the synthesis of SPIN-1 as a porous organic material, making use of the hydrolysis of multi-armed triazine with imidazolium followed by assembly of the ionic segments.
Results and discussion
In order to construct SPIN-1, we first synthesized 2,4,6-tri(1H-imidazol-1-yl)-1,3,5-triazine (2, Scheme 1) in 71% yield and grams scale by modifying a reported procedure.40,41 The structure 2 was characterized by FT-IR (Fig. 3) and 1H-NMR (Fig. S1†). The adsorption peaks at 3143 and 3113 cm−1 indicated the presence of unsaturated C–H bond, i.e. from imidazole. Three peaks at 7.25, 8.34 and 9.08 ppm in 1H-NMR revealed the presence of only one kind of imidazole ring. The existence of 2 was confirmed by HR-ESI-MS (Fig. S2†) based on [M + H]+ peak at 280.1052, which was required to be 280.1014. The above results were further supported by its elemental analysis, showing C 51.63%, H 3.12%, N 44.95% (Table S2†). The efforts to make SPIN-1 initiated by mixing cyanuric chloride and compound 2 at room temperature and in acetonitrile, which led to orange participation immediately. Considering the hydrolysis of positively charged triazine will release negative/positive charges and help the assembly via electrostatic interaction, we stir the reaction mixture in water/acetonitrile for 2 h. To our delight, the sample of SPIN-1-1 gave the Brunauer–Emmett–Teller (BET) surface area of 109 m2 g−1 (entry 1, Table 1). By lowering the concentration of 2 from 50 wt% to 1.5 wt%, the BET surface area increased to more than 263 m2 g−1 (entry 2, Table 1), with the increase of pore volume from 0.37 to 0.75 m3 g−1. However, the efforts to increase either the assembly temperature or drying temperature lead to lower BET surface (entry 3 and 4, Table 1). This shows the mild condition is suitable for the preparation of SPIN-1 with high BET surface.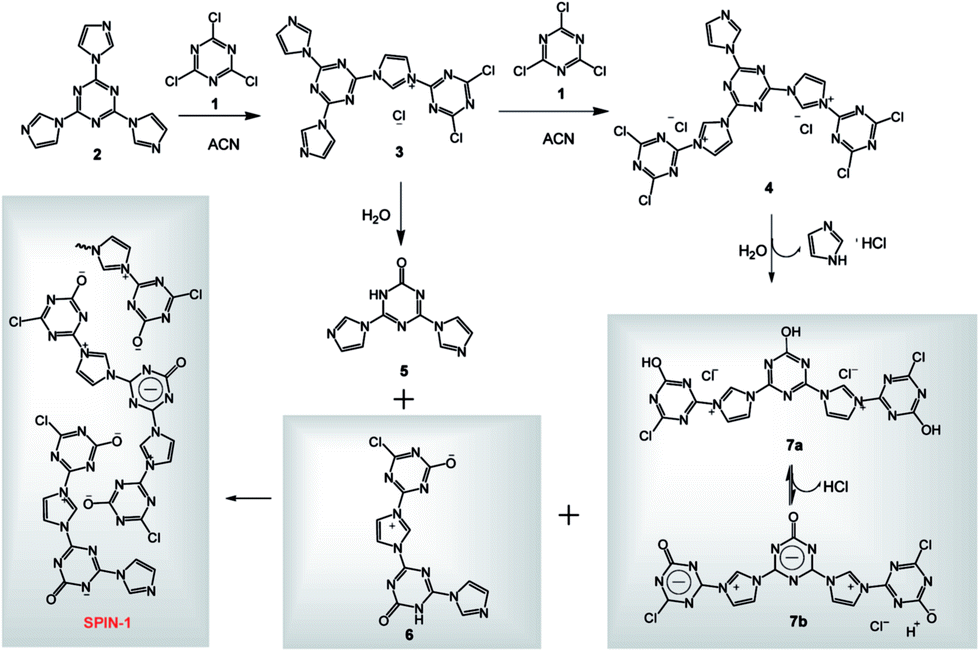 | ||
| Scheme 1 Possible structure and formation mechanism of SPIN-1. | ||
 | ||
| Fig. 2 (a) Adsorption (filled symbols) and desorption (empty symbols) N2 isotherm of SPIN-1-2 at 77 K; (b) pore size distribution curve by DFT. | ||
 | ||
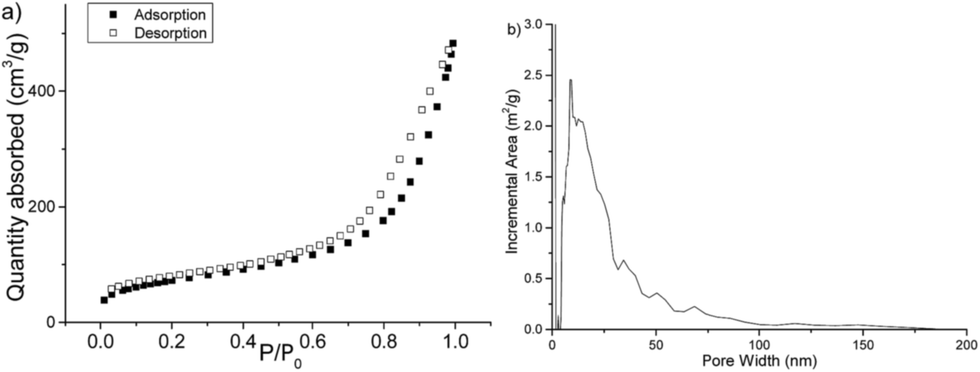
| Fig. 3 FT-IR spectra of 2 (blue) and SPIN-1 (red). | ||
| Entry | Conc. of 2 (wt%) | Assembly temp.a | Drying temp.b | SAc/m2 g−1 |
|---|---|---|---|---|
| a The temperature for stirring the mixture with H2O before filtration.b The temperature to remove residual solvent from the materials.c The BET surface area.d No assembly process was involved. | ||||
| 1 | 50 | r.t. | r.t. | 109 |
| 2 | 1.5 | r.t. | r.t. | 263 |
| 3 | 1.5 | r.t. | 85 °C | 152 |
| 4 | 1.5 | 90 °C | r.t. | 154 |
| 5 | 1.5 | NAd | r.t. | 40 |
The use of solvents is reported to be crucial to make porous polymerised ionic liquids.32 In our case, the role of water here is surely to help hydrolysis of positively-charged triazine. In addition, it acts as good solvent for the assembly and poor solvent to dissolve the as-prepared SPIN-1. As a control experiment, the solid obtained from mixing 1 and 2 was collected by filtration, but without the hydrolysis and assembly process (entry 5, Table 1). Indeed, the SPIN-1-5 was measured to have surface area as low as 40 m2 g−1.
The isotherm of SPIN-1-2 with BET surface of 263 m2 g−1 was shown in Fig. 2. The increase in the nitrogen sorption at a high relative pressure above 0.8 as well as a hysteresis loop at a high relative pressure (0.6 < P/P0 < 1.0) indicated a meso/macroporosity. Pore distribution calculated by DFT (Fig. 2) revealed an average pore size of 11 nm. BJH calculation shows 1.4% of them are micropores, 77% of them are mesopores, and 21% of them are macropores (Fig. S4†). Although SPIN-1 has a wide pore size distribution (Fig. 2b), all samples prepared (Table S1†) showed their pore volumes increased along the surface area.
With the sample of SPIN-1 with high BET surface area (entry 2 of Table 1), structure characterizations on it were carried out based on FT-IR, solid NMR, elemental analysis (EA), X-ray photoelectron spectra (XPS) and so on. The comparison between the IR spectra of 2 and SPIN-1 was shown in Fig. 3. The absorption peak at 3115 cm−1 in compound 2 (C–H of imidazole ring) has moved to higher wavenumbers in SPIN-1, indicating the formation of charged imidazole rings. The skeleton absorption at 810 cm−1 moved to lower wavenumbers in SPIN-1, indicating that the triazine rings were in negatively charged state (Scheme 1).
The absorption peaks for both SPIN-1 and compound 2 in the range of 1300–1600 cm−1 as well as 793–810 cm−1 are super similar, implying the main skeleton of imidazole and triazine remained unchanged. The absorption peaks at 1706 cm−1 in the FT-IR spectra of SPIN-1 (Fig. 3, red line) indicated the presence of carbonyl group, which did not exist in compound 2 (Fig. 3, blue line). However, this peak was very weak compared with peaks in the range of 1300–1500 cm−1, meaning the extent of carbonyl group is low, probably the phenol form is more favoured (Scheme 1). This comparison revealed clearly that the carbonyl group in SPIN-1 resulted from the hydrolysis of a triazine imidazolium intermediates. Beside FT-IR, the existence of triazine and imidazole subunit in SPIN-1 was characterized by CP TOSS 13C-NMR and was compared with 13C-NMR of compound 2 as shown in Fig. 4.
 | ||
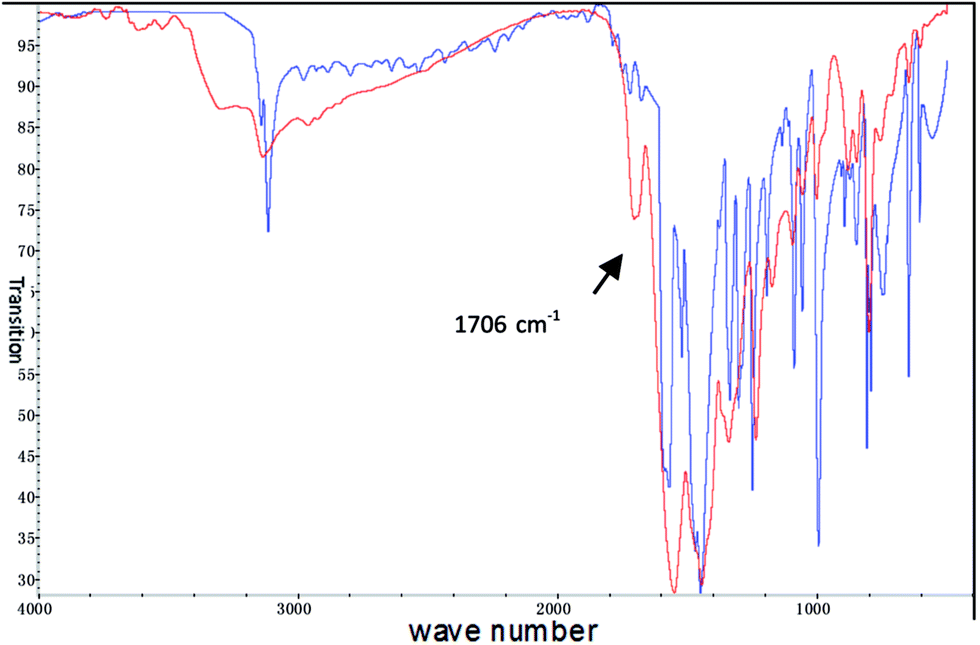
| Fig. 4 13C-NMR of 2 (red) and CP TOSS 13C-NMR of SPIN-1 (blue). | ||
The peaks for the carbon of triazine in 13C-NMR should appear at around 160 ppm as is detected in compound 2. However, four peaks were seen in the CP TOSS 13C-NMR in the range of 150–170 ppm, indicating the original neutral triazine ring was both activated as positively charged and deactivated as negatively charged. And the carbon of the carbonyl group resulted from hydrolysis of chlorotriazine shows the peak overlapped in this area. Similarly, the imidazole subunit shows several peaks in the range of 100–140 ppm, revealing neutral and positively charged imidazolium coexist in SPIN-1. The alternative triazononide and imidazolium conjugated structure was further supported by carefully comparing 13C-NMR of SPIN-1 with triazinonide bridged bisimidazolium salt.37
The structure of SPIN-1 was further characterized by X-ray photoelectron spectra (XPS, Fig. 5). Consistent with the result concluded from FT-IR, only a part of triazine rings were hydrolysed to give triazine-one (nitrogen at BE = 400.8 eV and oxygen at 531.5 eV) by XPS. Peaks at 400.8, 399.7 and 398.3 eV were assigned to be nitrogen atoms in hydrolysed triazine rings, imidazole rings and neutral triazine rings, respectively. The nitrogens didn't perform typical binding energy of ionic imidazole42 and triazines,43 which could be due to the counter ions linked directed to each other. It is also observed that the reaction between 1 and 3 followed by hydrolysis-assembly gave a quite acidic mixture (pH = 1), which provided white precipitates when treated with silver nitrate. This must come from HCl by hydrolysis of C–Cl bond in substituted chlorotriazine. However, after being washed twice by water, the lotion was neutral. Through washing, most free chlorides in SPIN-1 are gone, only small parts remained in the materials (200.0 and 201.7 eV). This could be the indirect evidence for the main chain alternative ionic structure.
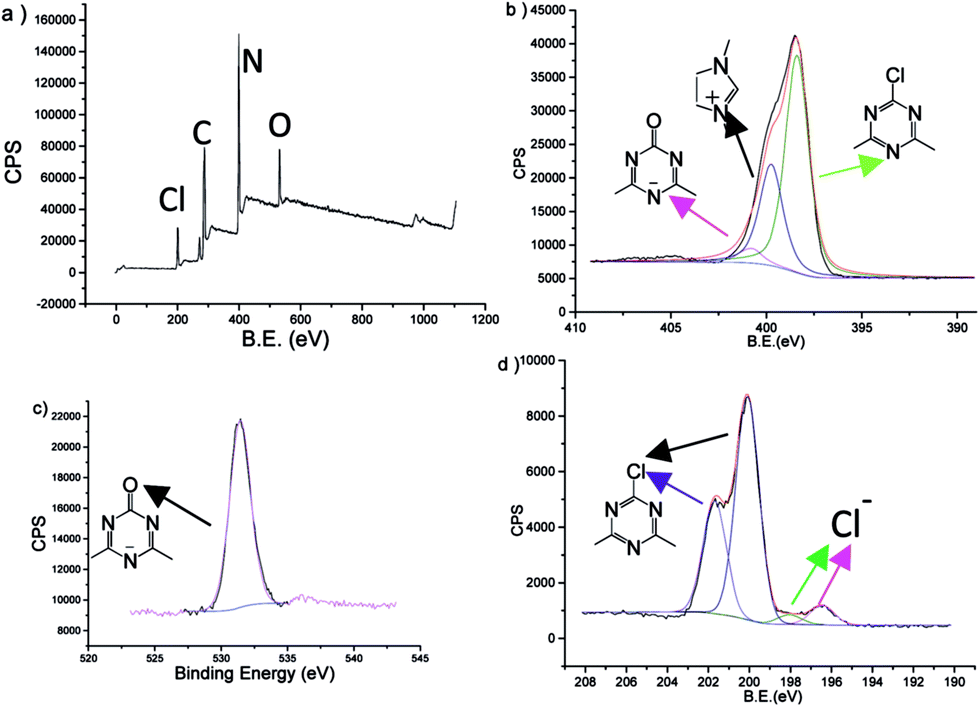 | ||
| Fig. 5 XPS data of SPIN-1: (a) survey; (b) N 1s (up); (c) O 1s; (d) Cl 2p. | ||
Based on the above observation, a possible chemical structure of SPIN-1 and assumed mechanism was shown in Scheme 1.
In acetonitrile, triimidazole triazine 2 reacted with cyacuric chloride 1 at room temperature to give imidazolium 3 and 4. The positively charged triazine ring in 3 and 4 became highly reactive and some of them could be hydrolyzed in the presence of water readily. For example, 3 could be hydrolyzed to give 5 and 6. 4 could be hydrolyzed to afford 7a, which could isomerize to 7b easily. These hydrolyzed chemicals with charges could assembly through electrostatic interaction (or ionic complexation) as well as π–π stacking to form the SPIN-1. These intermolecular interactions promote the formation of ionic cross-linking structure, which is non-equilibrium. This process force their chain conformation fixed to some extent. And this conformation fixation by ionic cross-linking is one of the reasons to create porosity. Once the supramolecular ionic network was formed, it precipitated as nanoparticles from water/acetonitrile phase owing to poorer solubility. And the nanoparticles further aggregate through supramolecular interaction to the final product of SPIN-1.
The morphology of SPIN-1 was observed by Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) (Fig. 6).
 | ||
| Fig. 6 Typical images of SPIN-1: (a) SEM of SPIN-1-5; (b) SEM of SPIN-1-2; (c) TEM of SPIN-1-2. | ||
The SEM for the sample SPIN-1-5 (prepared from reaction between compound 1 and 2 without hydrolysis and assembly procedure) shows it had typical particle accumulation morphology (Fig. 6a). The irregular particles (1–2 μm) packed closely to form irregular holes among them, leading to a poor porosity. As a comparison, a network structure at nano-micrometre scope with massive hollows (Fig. 6b) was found in the sample (SPIN-1-2) by in situ hydrolysis-assembly procedure. Macropores scaled from tens of nanometres to several micrometres could be observed directly, and the biggest pores were beyond the measurement range of gas adsorption. Through TEM image (Fig. 6c), hierarchical mesopores and macropores (shadows over 50 nm) can be observed, agreeing with the result from nitrogen sorption. It is clear to see from these pictures that SPIN-1 was a non-crystalline material, which can also be found through powder X-ray diffraction (PXRD) data (Fig. S3†). This could attribute to its fast formation while water was added in. It indicates that the self-assembly procedure could happen in a short time which make it hard to form ordered structure. It can be speculated that SPIN-1 formed its pores by a network at larger scale, in which the building units were not molecules or segments but molecular clusters linked by supramolecular interactions. SPIN-1 has a hierarchical meso/macroporosity like some composite materials, which is uncommon for typical porous organic polymers.
Climate change has become one major threat to the global ecosystem and human society. Carbon capture and storage (CCS) technology44 has been concerned to hold great promise as an alternative solution, and a variety of advanced porous materials45 have been developed for their potential application in CCS. As a new kind of charged porous organic polymers, Poly ionic liquids (PILs) have shown promising carbon dioxide capture capacity.46 Considering its ionic character similar to PILs, we investigated SPIN-1 toward CO2 capture capacity, which was measured by isotherm at 273 K (Fig. 7).
 | ||
| Fig. 7 CO2 isotherm for SPIN-1 at 273 K. | ||
It is up to 24.7 cm3 g−1 at 1 bar and 273 K, which is high among all PILs.47 While some of PILs shows open adsorption–desorption cycle, the adsorption of CO2 in SPIN-1 is reversible. The heat of adsorption of SPIN-1 was measured to be 22.4 kJ mol−1, which is also an evidence of physical adsorption (much less than PILs, up to 40 kJ mol−1). The interaction between CO2 molecules and SPIN-1 surface was not as strong as PILs, which could be a result of less activated imidazole ions that are not likely to form carbene. However, the combination of basic nitrogen sites and polar surface could still be effective, making SPIN-1 more suitable for CCS rather than chemical fixation.
Experimental
Materials
Imidazole and cyanuric chloride were purchased from Sinopharm Chemical Regent Co. N,N-Diisopropylethylamine (DIPEA) was purchased from Sigma Aldrich, tetrahydrofuran and acetonitrile were purchased from Beijing Tongguang Fine Chemical Company. All chemicals were used as received.Instrumentation
Liquid 1H NMR spectra were recorded in DMSO-D6 on Varian INOVA-400M magnetic resonance spectrometer. Solid-state NMR experiments were performed on a Bruker WB Avance II 400 MHz spectrometer. The 13C CP/MAS NMR spectra were recorded with a 4 mm double-resonance MAS probe and with a sample spinning rate of 12.0 kHz. FT-IR spectra of the samples were collected on a Nicolet 8700 Fourier transform spectrometer. The elemental analysis of the polymer was determined using aElementar Vario EL CUBE analyzer. The surface areas were screened using a Micromeritics ASAP 2020 volumetric adsorption analyzer with a 9-point BET measurement between the pressure range of 0.01–0.20 P/P0. Pore size distributions and pore volumes were derived from the adsorption branches of the isotherms using the density functional theory (DFT) pore model for pillared clay with cylindrical pore geometry. Samples were degassed for 12 h under vacuum before analysis. Carbon dioxide isotherms were measured at 273 K and 298 K using a Micromeritics ASAP 2020 volumetric adsorption analyzer. Field emission scanning electron microscopy (SEM) observations were performed on a Hitachi S-4800 microscope operated at an accelerating voltage of 10.0 kV. Transmission electron microscopy (TEM) images were obtained with a Tecnai G2 F30. Transmission electron microscope operated at 300 kV.Synthesis of 2,4,6-tri(1H-imidazol-1-yl)-1,3,5-triazine (2)
To a solution of imidazole (5.45 g, 80 mmol, 5 eq.) and N,N-diisopropylethylamine (DIPEA) (30 mL, 163 mmol, 10 eq.) in THF (40 mL) was added a solution of 1 (3.0 g, 16 mmol, 1 eq.) in THF (40 mL) portion wise at room temperature. The resulting mixture was stirred at room temperature for another 1 h, and some pale solid precipitated, which was washed by water followed by ethanol. After being dried at 85 °C under vacuum, the title compound was obtained as a white powder (3.2 g, yield 71%).IR (KBr, cm−1): 3143 (w), 3113 (w), 1572 (s), 1522 (m), 1448 (s), 1336 (m), 1303 (m), 1249 (s), 1194 (m), 1190 (m), 1057 (m), 996 (s), 895 (w), 850 (w), 810 (s), 794 (m), 743 (m), 648 (s), 607 (w), 558 (w).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.25 (s, 3H), 8.34 (s, 3H), 9.08 (s, 3H).
13C NMR (400 MHz, DMSO-d6) δ (ppm): 117.6, 131.1, 137.7, 162.1.
HR-MS (ESI): m/z 280.10522 [M + H]+ (found C12H10N9+, required 280.1014).
Elemental analysis: C 51.63%, H 3.12%, N 44.95% (theoretical value C 51.61%, H 3.25%, N 45.14%).
Synthesis of SPIN-1
To a solution of 2 (150 mg, 0.54 mmol, 1 eq.) in acetonitrile (100 mL) was added a solution of 1 (100 mg, 0.54 mmol, 1 eq.) in acetonitrile (5 mL). After stirred for 2.5 h, some orange solid was formed. The reaction system was allowed to open and water (150 mL) was added in. The resulting mixture was stirred at room temperature for another 2 h, and the solid became pale orange, which was flittered out and washed by water followed by ethanol for three times separately. Drying at room temperature under vacuum gave pale yellow powder of 70 mg.IR (KBr, cm−1): 3137 (w), 1706 (w), 1550 (s), 1444 (s), 1341 (m), 1236 (m), 1174 (m), 1058 (w), 1003 (w), 881 (w), 849 (w), 801 (m).
Elemental analysis: C 38.89%, H 2.79%, N 35.44%.
Conclusions
In summary, a template-free, robust and cost effective strategy has been developed to construct a supramolecular porous ionic network with alternative triazine and imidazole ions. The structure of SPIN-1 was fully characterized, and the mechanism to form its structure and porosity was proposed. Nitrogen sorption isothermal exhibits that SPIN-1 processes BET surface area up to 263 m2 g−1, with hierarchical meso-/macroporosity. As a result of its ionic porous surface, SPIN-1 has a carbon dioxide capture capacity up to 24.7 cm3 g−1, which is competitive among ionic porous organic polymers. Further application of SPIN-1 on gas adsorption, catalysis and solid conductor are under investigation.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21202008) and Beijing Natural Science Foundation (No 2162039) for generous support.Notes and references
- Q. Chen, M. Luo, P. Hammershoej, D. Zhou, Y. Han, B. W. Laursen, C. Yan and B. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- X. Lu, D. Jin, S. Wei, Z. Wang, C. An and W. Guo, J. Mater. Chem. A, 2015, 3, 12118–12132 CAS.
- C. Pei, T. Ben and S. Qiu, Mater. Horiz., 2015, 2, 11–21 RSC.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. I. Goddard, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- Q. Sun, Z. Dai, X. Meng, L. Wang and F. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS.
- S. Ding, J. Gao, Q. Wang, Y. Zhang, W. Song, C. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- R. Rohan, K. Pareek, Z. Chen, W. Cai, Y. Zhang, G. Xu, Z. Gao and H. Cheng, J. Mater. Chem. A, 2015, 3, 20267–20276 CAS.
- J. F. Van Humbeck, M. L. Aubrey, A. Alsbaiee, R. Ameloot, G. W. Coates, W. R. Dichtel and J. R. Long, Chem. Sci., 2015, 6, 5499–5505 RSC.
- Q. Zhao, J. W. C. Dunlop, X. Qiu, F. Huang, Z. Zhang, J. Heyda, J. Dzubiella, M. Antonietti and J. Yuan, Nat. Commun., 2014, 5, 4293 Search PubMed.
- Y. Yuan, F. Sun, L. Li, P. Cui and G. Zhu, Nat. Commun., 2014, 5, 4260 RSC.
- S. Zhang, K. Dokko and M. Watanabe, Chem. Sci., 2015, 6, 3684–3691 RSC.
- S. Mitra, S. Kandambeth, B. P. Biswal, A. M. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed.
- J. F. Van Humbeck, T. M. McDonald, X. Jing, B. M. Wiers, G. Zhu and J. R. Long, J. Am. Chem. Soc., 2014, 136, 2432–2440 CrossRef CAS PubMed.
- Y. Yuan, F. Sun, F. Zhang, H. Ren, M. Guo, K. Cai, X. Jing, X. Gao and G. Zhu, Adv. Mater., 2013, 25, 6619–6624 CrossRef CAS PubMed.
- J. S. Lee, H. Luo, G. A. Baker and S. Dai, Chem. Mater., 2009, 21, 4756–4758 CrossRef CAS.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- K. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C. Sue, T. Zhou, L. Zhang, X. Zhao, Y. Liu and Z. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef CAS PubMed.
- J. Tian, T. Zhou, S. Zhang, S. Aloni, M. V. Altoe, S. Xie, H. Wang, D. Zhang, X. Zhao, Y. Liu and Z. Li, Nat. Commun., 2014, 5, 5574 CrossRef CAS PubMed.
- M. Pfeffermann, R. Dong, R. Graf, W. Zajaczkowski, T. Gorelik, W. Pisula, A. Narita, K. Muellen and X. Feng, J. Am. Chem. Soc., 2015, 137, 14525–14532 CrossRef CAS PubMed.
- M. Wathier and M. W. Grinstaff, J. Am. Chem. Soc., 2008, 130, 9648–9649 CrossRef CAS PubMed.
- G. Godeau, L. Navailles, F. Nallet, X. Lin, T. J. McIntosh and M. W. Grinstaff, Macromolecules, 2012, 45, 2509–2513 CrossRef CAS PubMed.
- M. A. Aboudzadeh, M. E. Munoz, A. Santamaria and D. Mecerreyes, RSC Adv., 2013, 3, 8677–8682 RSC.
- M. A. Aboudzadeh, A. S. Shaplov, G. Hernandez, P. S. Vlasov, E. I. Lozinskaya, C. Pozo-Gonzalo, M. Forsyth, Y. S. Vygodskii and D. Mecerreyes, J. Mater. Chem. A, 2015, 3, 2338–2343 CAS.
- R. Rohan, Y. Sun, W. Cai, K. Pareek, Y. Zhang, G. Xu and H. Cheng, J. Mater. Chem. A, 2014, 2, 2960–2967 CAS.
- S. Chakraborty, Y. J. Colon, R. Q. Snurr and S. T. Nguyen, Chem. Sci., 2015, 6, 384–389 RSC.
- Q. Zhao, P. Zhang, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2012, 134, 11852–11855 CrossRef CAS PubMed.
- S. Chakraborty, Y. J. Colon, R. Q. Snurr and S. T. Nguyen, Chem. Sci., 2015, 6, 384–389 RSC.
- J. H. Lee, H. J. Lee, S. Y. Lim, B. G. Kim and J. W. Choi, J. Am. Chem. Soc., 2015, 137, 7210–7216 CrossRef CAS PubMed.
- G. Ji, Z. Yang, H. Zhang, Y. Zhao, B. Yu, Z. Ma and Z. Liu, Angew. Chem., Int. Ed., 2016, 55, 9685–9689 CrossRef CAS PubMed.
- Q. Sun, S. Ma, Z. Dai, X. Meng and F. Xiao, J. Mater. Chem. A, 2015, 3, 23871–23875 CAS.
- C. Gao, H. Zhou, S. Wei, Y. Zhao, J. You and G. Gao, Chem. Commun., 2013, 49, 11820 CAS.
- H. Zhou, Y. Zhao, G. Gao, S. Li, J. Lan and J. You, J. Am. Chem. Soc., 2013, 135, 14908–14911 CrossRef CAS PubMed.
- H. Zhou, Z. Wang, C. Gao, J. You and G. Gao, Chem. Commun., 2013, 49, 1832–1834 RSC.
- X. Gan, L. Kong, P. Wu, C. Lv, Y. Tu, Y. Chen, H. Zhou, J. Wu and Y. Tian, Chin. J. Struct. Chem., 2011, 30, 1650–1655 CAS.
- D. Azarifar, M. A. Zolfigol and A. Forghaniha, Heterocycles, 2004, 63, 1897–1901 CrossRef CAS.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H. Steinrück, Chem.–Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed.
- Y. Zhang, L. Hu, C. Zhu, J. Liu, H. Huang, Y. Liu and Z. Kang, Catal. Sci. Technol., 2016 10.1039/C6CY01066K.
- Y. Tan, W. Nookuea, H. Li, E. Thorin and J. Yan, Energy Convers. Manage., 2016, 118, 204–222 CrossRef CAS.
- G. Ferey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. De Weireld, A. Vimont, M. Daturi and J. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC.
- A. Wilke, J. Yuan, M. Antonietti and J. Weber, ACS Macro Lett., 2012, 1, 1028–1031 CrossRef CAS.
- S. Zulfiqar, M. I. Sarwar and D. Mecerreyes, Polym. Chem., 2015, 6, 6435–6451 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures. See DOI: 10.1039/c6ra20590a |
| This journal is © The Royal Society of Chemistry 2016 |