DOI:
10.1039/C6RA20568B
(Paper)
RSC Adv., 2016,
6, 99414-99421
High-sensitivity stimuli-responsive polysiloxane synthesized via catalyst-free aza-Michael addition for ibuprofen loading and controlled release
Received
15th August 2016
, Accepted 13th October 2016
First published on 13th October 2016
Abstract
Herein, we report a strategy to synthesize a series of high-sensitivity stimuli-responsive polysiloxanes (SPSis) via a facile, highly efficient, catalyst-free aza-Michael addition of poly(aminopropylmethylsiloxane) (PAPMS) with N-isopropylacrylamide. The SPSis structures are systematically characterized using FT-IR, 1H NMR and 13C NMR. The effects of N-isopropyl amide group (NIPAs) content in PAPMS, pH, and salinity on the responsive properties of SPSis are examinated in detail. The as-prepared SPSis show high sensitive respond to three changes of thermo-, pH-, and salinity, where the phase separation occurred within 0.3 °C due to their flexible Si–O–Si backbone, and lower critical phase separation (LCST) changed from 14.7 °C to 57.0 °C at varied NIPAs contents. Surprisingly, the SPSis exhibit effectively hydrogen bond-derived loading (up to 74 wt%) and temperature-dependent release of hydrophobic drugs (ibuprofen). The present methodology may open a route for developing high-sensitive multi-stimuli-responsive polymers.
1. Introduction
Stimuli-responsive polymers exhibit rapid, reversible, and dramatic changes in physical and chemical properties (e.g. hydrophilic and hydrophobic, solubility, conformation, and shape) in response to external stimuli.1 Therefore, those polymers exhibit unique advantages in molecular filtration,2 sensors,3 bioimaging,4 cell culture,5 drugs and bio-molecules delivery.6–10 Recent studies have reported smart copolymers based on poly(N-isopropylacrylamide) [poly(NIPAM)].11–15 However, improving the sensitiveness of these materials is complicated, and those polymers with acrylic acid or acrylamide display a limited lower critical solution temperature (LCST) range, slow response with observable hysteresis and high cytotoxicity to external stimuli.16–18 Polymer backbones play an important role in phase transition because the process is derived from the transformation of the polymer conformation from an expanded form to a condensed one, dependent on the conformation energy of the backbone.19 Therefore, we speculate that smart polymers with the Si–O–Si backbone should be more sensitive to environmental changes than the ones with the C–C backbone, because of the repeated highly flexible Si–O units.
Polysiloxane is an appealing material for clinical and medical treatments, because of its outstanding physiological inertness, non-toxicity, and biocompatibility.20–23 However, traditional polysiloxanes show poor hydrophilicity and insensitivity to the environment, which limit the use of the materials in smart fields. Currently, polysiloxanes with stimuli-responsive abilities are synthesized by modification polysiloxane matrix with smart polymers or changing polysiloxane into amphipathic block copolymers.24–26 However, polysiloxane segments in these polymers merely serve as accessories and show no stimuli-responsive capacities. To the best of our knowledge, stimuli-responsive polysiloxanes based on the Si–O–Si backbone have not yet been synthesized because of the problem in combining NIPAM and polysiloxane. Poly(NIPAM)-based polymers are synthesized by complex and elaborate atom transfer radical polymerization27,28 or reversible addition–fragmentation chain transfer.29,30 Hydrolytic polycondensation and ring-opening polymerization are the main methods in polysiloxane preparation. Hydrosilylation involving a Si–H bond across a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond is an important reactions in silicone chemistry.31 However, platinum-based noble metal catalysts, which are generally obligatory, are easily deactivated once in the presence of NH2 or OH groups. Furthermore, the selectivity of the Pt-catalyzed hydrosilylation of allyl derivatives is low because the formal Tsuji–Trost reaction is competitive.32
C double bond is an important reactions in silicone chemistry.31 However, platinum-based noble metal catalysts, which are generally obligatory, are easily deactivated once in the presence of NH2 or OH groups. Furthermore, the selectivity of the Pt-catalyzed hydrosilylation of allyl derivatives is low because the formal Tsuji–Trost reaction is competitive.32
In the current study, we present the SPSis with N-isopropyl amides (NIPAs) as side groups and polysiloxane as backbone synthesized by aza-Michael addition, namely the reaction of activated alkenes (α,β-unsaturation carbonyl compound) and amines. In specific, the reaction yields no by-products and exhibits high function group tolerance and high conversions at room temperature.33–36 The condition provides a convenient and practical method of SPSis synthesis. Compared with PNIPAM-based smart polymers, the as-prepared SPSis are more sensitive to temperature, pH, and salinity, and own a broad controllable LCST range. SPSis with biocompatibility and biodegradability exhibit a great hydrogen bond-derived loading and temperature-dependent release of hydrophobic drugs. This mechanism is one of the surprising characteristics of SPSis. We selected ibuprofen, a nonsteroidal anti-inflammatory drug widely used as analgesic and antipyretic, as a model to evaluate the ability of the SPSis.
2. Experimental
2.1 Materials
1,3-Bis(3-aminopropyl)-1,1,3,3-tetramethyldisiloxane (AP2-PSi2) and aminopropylmethyldiethoxysilane (APMDES) of industrial grade were obtained from Shenzhen Changhao Technology Company and used after distillation under reduced pressure and determined by 1H NMR. NIPAM and Ibuprofen of analytically pure were obtained from Energy Chemical China. 2,4,6,8-Tetraaminopropyl-2,4,6,8-tetramethyl cyclotetrasiloxane (D4-AP) was prepared by a hydrolysis-cracking process of APMDES as follows. APMDES of 100 g, toluene of 100 g, H2O of 50 g, and KOH of 1 g were added in a round bottom flask. The mixture was refluxed for 30 min and then distilled slowly to remove volatiles under atmospheric pressure and colourless viscous product was obtained after vacuum distillation at 2 mm Hg and 220 °C.
2.2 Synthesis of SPSis
As shown in Scheme 1, poly(aminopropylmethylsiloxane) (PAPMS) was synthesized by the ring-opening polymerization of D4-AP using KOH as a catalyst, DMSO as an accelerator and AP2-PSi2 as an end-capping reagent. Typically, 23.4 g (0.05 mol) D4-AP was injected with nitrogen into a three-neck flask equipped with a mechanical stirrer. Then, 0.047 g (0.2 wt%) KOH and 0.23 g (1.0 wt%) DMSO was added to the mixture, and the temperature was increased to 50 °C. The viscosity of the reaction mixture sharply increased after an hour or so. 1.25 g (0.005 mol) AP2-PSi2 was added rapidly to the flask, and the reaction was equilibrated for 5 h. PAPMS was obtained after the mixture cooled to room temperature and neutralized by an equimolar acetic acid with KOH. Gel permeation chromatography shows the as-prepared PAPMS with molar weight of 4980 g mol−1 (PDI: 1.82), about 42 Si–O units. Then, 2.5 g PAPMS and NIPAM of a designed mass (0.48 g, 0.97 g, 1.45 g, 1.93 g and 2.42 g) were dissolved in 25 mL ethanol, and stirred for 48 h at 30 °C. SPSis were obtained after the ethanol was removed by rotary evaporation and vacuum drying. A viscous product was obtained. The NIPAs content in SPSi was controlled by the feed molar ratio of NIPAM in system.
 |
| | Scheme 1 Synthesis of SPSis via catalyst-free aza-Michael addition. | |
2.3 LCST characterization of SPSis
The LCST of SPSis in water (10 mg mL−1) was recorded by a Tu-1901 UV-vis spectrophotometer equipped with a DKB-1906 incubator at 500 nm. To obtain temperature-dependent spectra, the initial temperature was kept at 10 °C and increased to 60 °C at a rate of 1.0 °C min−1. The mixture was a transparent solution at 10 °C and set as 100% transmittance. The temperature-dependent spectra of various pH, and salinity were obtained through the similar method. The pH value of system was adjusted by addition of HCl or NaOH solution (0.1 mol L−1).
2.4 Chemical structure
The chemical structure of SPSis was characterized by FT-IR using a Bruker TENSOR 27 instrument within the range of 4000–400 cm−1 working at 4 cm−1 resolution. 1H NMR and 13C NMR spectra were recorded in CDCl3 using a Bruker 300 MHz instrument. The conversion of NIPAM into SPSis was determined from the characteristic signals of –Si–CH2– (at 0.4 ppm) and –NH–CH(CH3)2 (at 1.0 ppm) and the molar ratios of mono-addition and bis-addition were obtained from the characteristic signals of –NH–CH2– (at 2.8 ppm) and –N–[(CH2)–]2 (at 2.7 ppm). Gel permeation chromatography (GPC) measurements were performed on a Milford. MA waters 515 liquid chromatography equipped with a 2414 refractive-index detector. Samples were run in THF at 40 °C at a rate of 1 mL min−1.
2.5 Solubilisation and release of ibuprofen
Ibuprofen of 100 mg was added into 10 mL SPSis aqueous of varied concentrations (0–10 mg mL−1) in batches in ice water bath until the ibuprofen was no longer dissolved. Undissolved ibuprofen was removed by centrifugation, and the supernatant was diluted by PBS solution (0.02 mol L−1) and determined by UV-spectroscopy at 267 nm to get the solubility of ibuprofen at different conditions. The drug-loaded SPSis was put into a dialysis tube (Mw: 1500) and immerse into 20 mL PBS solution at different temperature. The soak solution was determined by UV-spectroscopy once per hour to calculate the released ibuprofen.
3. Results and discussion
3.1 Synthesis, design, and characterization of SPSi
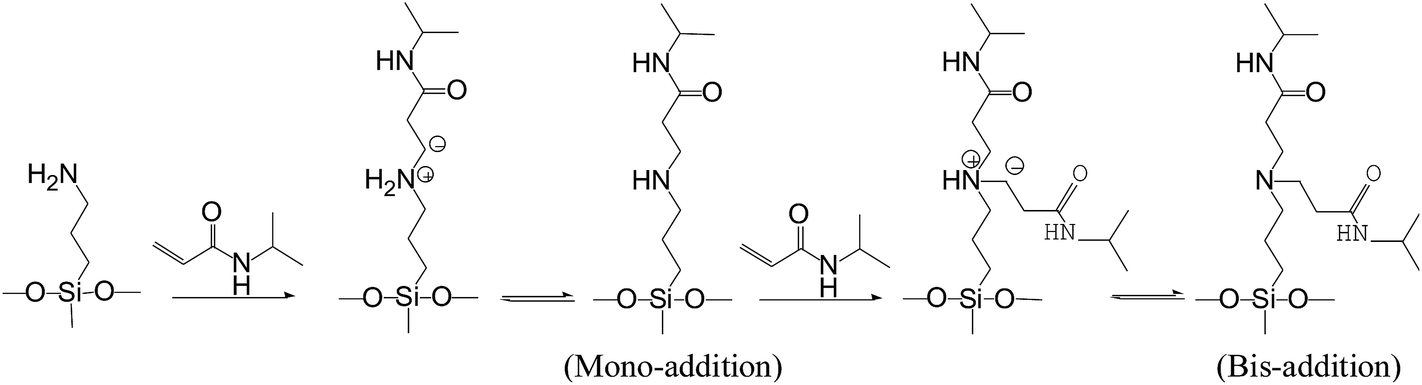
The SPSis were obtained from the post-functioned of PAPMS and NIPAM through the aza-Michael addition in ethanol at room temperature. The key of the addition depends on the active of nucleophilic substitution between amino groups in PAPMS and the double bond in NIPAM. The amines can either act as nucleophiles or bases, and NIPAM as olefin derivatives, possesses an electron-withdrawing activating group (CO), which stabilizes the resulting intermediate until proton transfer occurs (Fig. 1). Surprisingly, the addition could proceed at room temperature without any catalyst and initiator. Bis-addition is inevitable because imino groups in mono-addition product were more nucleophilic than primary amino. As shown in Table 1, bis-addition decreased from 14.46% to 7.98% when NIPAs contents decreased from 100% to 60%. When NIPAs contents were below to 40%, the characteristic peak of –CO–N–[(CH2)–]2 become indiscernible (Fig. 3), so bis-addition was ignored.
 |
| | Fig. 1 Mechanism of aza-Michael addition reaction of PAPMS with NIPAM. | |
Table 1 Aza-Michael addition dates and LCST of the SPSis
| Samples |
NIPAsa/% |
Addition type/% |
LCST/°C |
| Feed |
1H NMR |
Mono- |
Bis- |
| The content of NIPAs was calculated base on SI–O units. The peaks of bis-addition are indiscernible in 1H NMR. |
| SPSis-100 |
100 |
100.00 |
85.54 |
14.46 |
14.7 |
| SPSis-80 |
80 |
76.33 |
89.10 |
10.90 |
16.8 |
| SPSis-60 |
60 |
56.33 |
92.02 |
7.98 |
19.4 |
| SPSis-40 |
40 |
39.75 |
100 |
—b |
27.8 |
| SPSis-35 |
35 |
34.26 |
100 |
— |
32.7 |
| SPSis-30 |
30 |
29.91 |
100 |
— |
40.0 |
| SPSis-20 |
20 |
20.17 |
100 |
— |
57.0 |
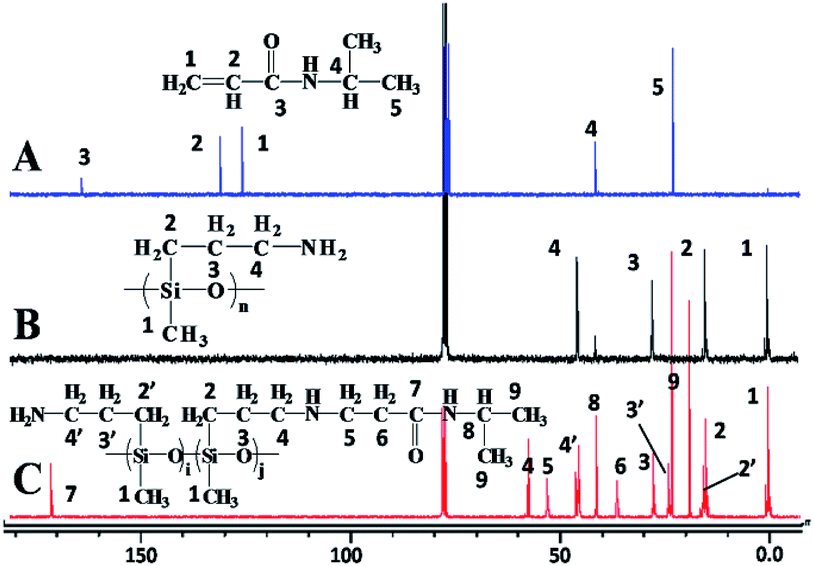
The 1H NMR of the monomers (NIPAM, PAPMS) and SPSis-40 are displayed in Fig. 2. The resonance peaks at 4.2, 6.1 and 6.2 ppm assigned to double bond (CH2CH–) of NIPAM (H1, H2 in Fig. 2A) disappeared in the SPSis (Fig. 2C).37 However, the newly formed –NH–CH2–CH2–CO– group displayed peaks at 2.2 and 2.8 ppm (H6, H7 in Fig. 2C), indicating that all of the CH2CH– groups of NIPAM reacted with amino groups. In addition, the peak of –CH3 group of NIPAM at 1.2 ppm appeared in SPSis (H10 in Fig. 2C), confirming the conversion of NIPAM to SPSis. Peaks of the –Si–CH3, –Si–CH2–, and –Si–CH2–CH2– at 0, 0.5, and 1.2 ppm (H1, H2, H3 in Fig. 2B and C), respectively, confirm the presence of alkylsilane groups in SPSis. In Fig. 3, we can clearly see that, the peak at 2.7 ppm belonged to bis-addition products (–N–(CH2–)2) decreased with the NIPAM feed molar ratios and disappeared when 40% of NIPAM added. The ratios of mono-addition/bis-addition were recorded by the ratio of peak 6/peak 6′ in Fig. 3 and listed in Table 1.
 |
| | Fig. 2 1H NMR spectra of NIPAM (A), PAPMS (B), and SPSis-40 (C). | |
 |
| | Fig. 3 The details of aza-Michael addition with varied NIPAM feed molar ratios. | |
13C NMR tests on SPSis along with NIPAM, APMDES and PAPMS were also carried out and the spectra are displayed in Fig. 4. Peaks at 136 and 130 ppm assigned to the double bond (CH2CH–) of NIPAM (C1, C2 in Fig. 4A) disappeared in the SPSis (Fig. 4C). The new formed –NH–CH2–CH2–CO– group exhibited peaks at 53 and 36 ppm (C5, C6 in Fig. 4C), indicating that the CH2CH– group of NIPAAM reacted with amino groups. In addition, the peaks of –CH3 and –CO groups of NIPAM at 24 ppm and 165 ppm (C5, C3 in Fig. 4A) appeared in SPSis (C9, C7 in Fig. 4C), confirming the conversion of NIPAM to SPSis. The characteristic peaks of –Si–CH3 and –Si–CH2– at 0.0, and 14.7 ppm, respectively, were exhibited by PAPMS (C1, C2 in Fig. 4B) and SPSis (C1, C2 in Fig. 4C, indicating the existence of alkylsilane groups in SPSis. Accordingly, the original peaks of –Si–CH2–CH2– and –Si–CH2–CH2–CH2– at 28 and 46 ppm in APMDES and PAPMS (C3, C4 in Fig. 4B), respectively, give two peaks at 58, 46 ppm and 25, 27 ppm (C4, C′4 and C3, C′3 in Fig. 4C), that contributed to the amino group partially reacted with NIPAM, giving different electronic environment of carbon atom adjacent to amino.
 |
| | Fig. 4 13C NMR spectra of NIPAM (A), PAPMS (B), and SPSis-40 (C). | |
FT-IR spectroscopy of monomers and SPSis-40 were compared in Fig. 5. Clearly visible singles at 3296 cm−1, 1659 cm−1 and 1549 cm−1 assigned to the stretching vibration of the NH group, stretching vibration of the CO group and bending vibration of the NH group in NIPAM were observed. These singles were also observed in NIPDES, and SPSis-40, indicating that NIPAM was introduced in SPSis successfully. However, the single at 1618 cm−1 assigned to conjugate stretching vibration CC in NIPAM disappeared in SPSis-40, which confirms that an aza-Michael addition reaction occurred between NIPAM and NH2. Singles at 1090 cm−1 and 797 cm−1 assigned to the Si–O–Si asymmetric bending, Si–O symmetric stretching mode, and Si–O–Si symmetric bending mode, respectively, were observed in SPSis-40, confirming the Si–O–Si backbone of SPSis. In Addition, broad singles from 3200 cm−1 to 3500 cm−1 in SPSis-40 are assigned to stretching vibration of the NH2 group. The adsorption at 3072 cm−1 belongs to the overtone bands of stretching vibration of NH. Singles from 2800 cm−1 to 2980 cm−1 are assigned to the vibration of –CH3 and –CH2 groups. Single bands at 1258 cm−1 are belong to the common vibration features of the Si–C groups.
 |
| | Fig. 5 FT-IR spectra of NIPAM (A), PAPMS (B), and SPSis-40 (C). | |
As-prepared SPSis could dissolve in water at low temperature, and precipitate to form polymersomes on heating over the LCST (Fig. 6) (the temperature of each sample discussed in later part). The solubility of SPSis in water originates from hydrogen bonds between the host framework with hydrophilic groups and hydrophobic interactions in SPSis. The polymer chains at low temperatures are in the energetically favored hydrated state, as water molecules associated with the amino groups form H-bonding cage-like structures around the hydrophobic isopropyl group. As the temperature increases, the hydrogen bonding becomes weaker. Above the LCST, intramolecular hydrogen bonds between CO and N–H groups in the chains result in a compact and hydrophobic collapsed conformation of polymer chains and assembled into polymersomes. Thus, the LCST of as-prepared SPSis are influenced by the conditions that influence hydrogen bond.
 |
| | Fig. 6 Phase separations of SPSis and the interplay of hydrophilic and hydrophobic. | |
3.2 The effects of NIPAs contents on LCST of SPSis
Fig. 7 shows the temperature dependence of optical transmittance of SPSis at pH = 7.0. Compared to common poly(NIPAM)-based smart polymers, an important feature of the SPSis is broad LCST crossed physiological conditions. As shown in Fig. 7, the LCST of SPSis exponentially increased from 14.7 °C to 57 °C, with the NIPAs molar ratios from 100% to 20.17%. This variation should be attributed to both the hydrophilic amino group and the hydrophilic/hydrophobic switchable NIPAs group. The LCST of a polymer in a given solvent may be adjusted in a wide range by the copolymerization of more hydrophobic or more hydrophilic monomers compared to the original main monomer.38 Hydrophobic co-monomers that favour segment–segment interactions in polymers decrease the LCST, whereas hydrophilic comonomers that favour segment–solvent interactions have the opposite affect.39–41 Copolymerization of NIPAM with acrylamide at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 co-monomer molar ratio results in the increase of copolymer LCST from 32 °C to 37.5 °C.42 Durmaz et al. reported star-shaped copolymers of hydrophobic hexyl methacrylate (HMA) and 2-(dimethylamino) ethyl methacrylate, when the ratios of hydrophobic HMA monomers were higher than 50 mole%, the polymer's aqueous solubility dramatically decreased.43 In the present study, unmodified amino displayed a hydrophilic comonomer effect that led to the increase of LCST, which is similar to results from Kumar's studies.44 When the molar ratio of NIPAs is 100%, the LCST of SPSis is 14.7 °C (SPSis-100), which is much lower than reported 32 °C of linear poly(NIPAM) homopolymers. This result is attributed to the more hydrophobic Si–O–Si backbone of SPSis. Phase transition needs a broad span of temperature, when 20% of NIPAs attached to Si–O–Si, the SPSis become insensitive to temperature. Once no NIPAs introduced, no phase transition can be observed (SPSis-0). This behavior is caused by the hydrophilic and hydrophobic display of NIPAs group in protonated or deprotonated forms, respectively, whereas the phase transition is attributed to the hydrophobicity of NIPAs group by deprotonation. Besides, even the NIPAs were reduced to 30%. The sudden phase transition of SPSis occurred in a range of 0.3 °C, which is much narrower than that of reported smart polymers.45 The high sensitivity comes from the soft Si–O–Si backbone of SPSis, which leads to a lower conformation energy required in a phase transition. Furthermore, the LCST regulation of SPSis rejected using of cytotoxic acrylamide.
:2 co-monomer molar ratio results in the increase of copolymer LCST from 32 °C to 37.5 °C.42 Durmaz et al. reported star-shaped copolymers of hydrophobic hexyl methacrylate (HMA) and 2-(dimethylamino) ethyl methacrylate, when the ratios of hydrophobic HMA monomers were higher than 50 mole%, the polymer's aqueous solubility dramatically decreased.43 In the present study, unmodified amino displayed a hydrophilic comonomer effect that led to the increase of LCST, which is similar to results from Kumar's studies.44 When the molar ratio of NIPAs is 100%, the LCST of SPSis is 14.7 °C (SPSis-100), which is much lower than reported 32 °C of linear poly(NIPAM) homopolymers. This result is attributed to the more hydrophobic Si–O–Si backbone of SPSis. Phase transition needs a broad span of temperature, when 20% of NIPAs attached to Si–O–Si, the SPSis become insensitive to temperature. Once no NIPAs introduced, no phase transition can be observed (SPSis-0). This behavior is caused by the hydrophilic and hydrophobic display of NIPAs group in protonated or deprotonated forms, respectively, whereas the phase transition is attributed to the hydrophobicity of NIPAs group by deprotonation. Besides, even the NIPAs were reduced to 30%. The sudden phase transition of SPSis occurred in a range of 0.3 °C, which is much narrower than that of reported smart polymers.45 The high sensitivity comes from the soft Si–O–Si backbone of SPSis, which leads to a lower conformation energy required in a phase transition. Furthermore, the LCST regulation of SPSis rejected using of cytotoxic acrylamide.
 |
| | Fig. 7 Temperature-dependent optical transmittance spectra of SPSis with various NIPAM feed ratios (A) and exponential relationship of NIPAs mole fractions and LCST of SPSis (B). | |
Because of the intra-molecular hydrogen bonding interaction, temperature induced phase separation is reversible and accompanied certain hysteresis.46 As shown in Fig. 8, narrow hysteresis between heating and cooling scans was observed. The NIPAs group decreased from 100% to 40%; NH2 group of SPSis increased from 0% to 60%, whereas the degree of hysteresis ΔLCST decreased from 1.3 °C to 0.7 °C. The hysteresis is due to the inter-molecular and intra-molecular hydrogen bonds, which hinder the rehydration process with decreasing temperature.47 Introducing water-soluble blocks or grafts into thermally responsive polymer backbones results in faster demixing–remixing cycle; this result is roughly about by the ability of water molecules to leave the polymer-rich phase and return, whereas its combination with hydrophobic monomer leads to an increased ΔLCST. Similarly, the presence of –NH2 groups in SPSis improves water molecules penetration in the polymer-rich phase. This behavior results in a smaller degree of thermal hysteresis (faster response) compared with other poly(NIPAM)-based smart polymers.
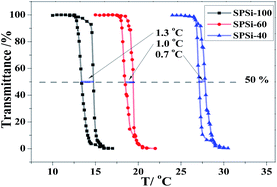 |
| | Fig. 8 Plots of the transmittance of SPSis aqueous solution (10 mg mL−1) as a role of temperature at a heating–cooling cycle at a rate of 0.5 °C min−1. | |
3.3 The effect of pH and salinity on phase transition behavior of SPSis
Double responsive copolymers can be synthesized by copolymerization of NIPAM with monomers have protonation/deprotonation ability, such as acrylic acid or acrylamide.48 Therefore; we examined the effect of pH value on the phase-transition behaviour of SPSis. As shown in Fig. 9, LCST of SPSis increased from 25.2 °C to 32.6 °C, whereas the pH value decreased from 9 to 4. The phase-transition occurs when the hydrogen bond breaks from the dehydrating solution and the polymer precipitates out. In the highly acidic solutions, the protonated amines resulted in electrostatic repulsion and expanded state; and the higher the hydrophilicity of the protonated amino, the higher the LCST was. At low pH, amino groups undergo ionization and improve polymer solubility, so the phase transition temperature increases to compensate for the enhanced hydrophilicity.44,49 Based on these considerations, the decrease of LCST in the basic SPSis solution upon heating is assumed to be a result of an increase in hydrophobicity driven compaction of the polymers.
 |
| | Fig. 9 Temperature-dependent optical transmittance spectra and LCST of SPSis (SPSis-40) at various pH values. | |
As shown in Fig. 10, SPSis are sensitive to the presence of electrolyte Na2SO4. When Na2SO4 concentration increased from 0 mol L−1 to 0.1 mol L−1, the LCST of SPSis dramatically decreased from 27.8 °C to 20.3 °C linearly (Fig. 10). This result agrees with previous research on the effect of salt on the solution properties of poly(NIPAM), which stated that increasing the salt concentration decreases the LCST of the polymer through the salting-out effect. For instance, the difference in LCST of poly(NIPAM) in water (25 mg mL−1) and in physiological saline (0.15 mol L−1) is 3 °C.50,51 The effect is attributed to the increase in polymer-water interfacial tension with the addition of salt, which favours intra-molecular hydrophobic interactions. Such response to changes in electrolyte concentration typically manifests as changes in conformation associated with hydrodynamic diameters of the polymers. Such behaviours are the polyelectrolyte and anti-polyelectrolyte effects.
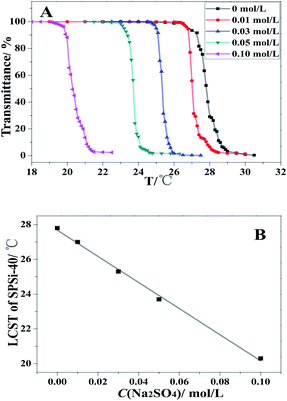 |
| | Fig. 10 Temperature-dependent optical transmittance spectra and LCST of SPSis (SPSis-40) at various salinities. | |
3.4 Loading and release of ibuprofen
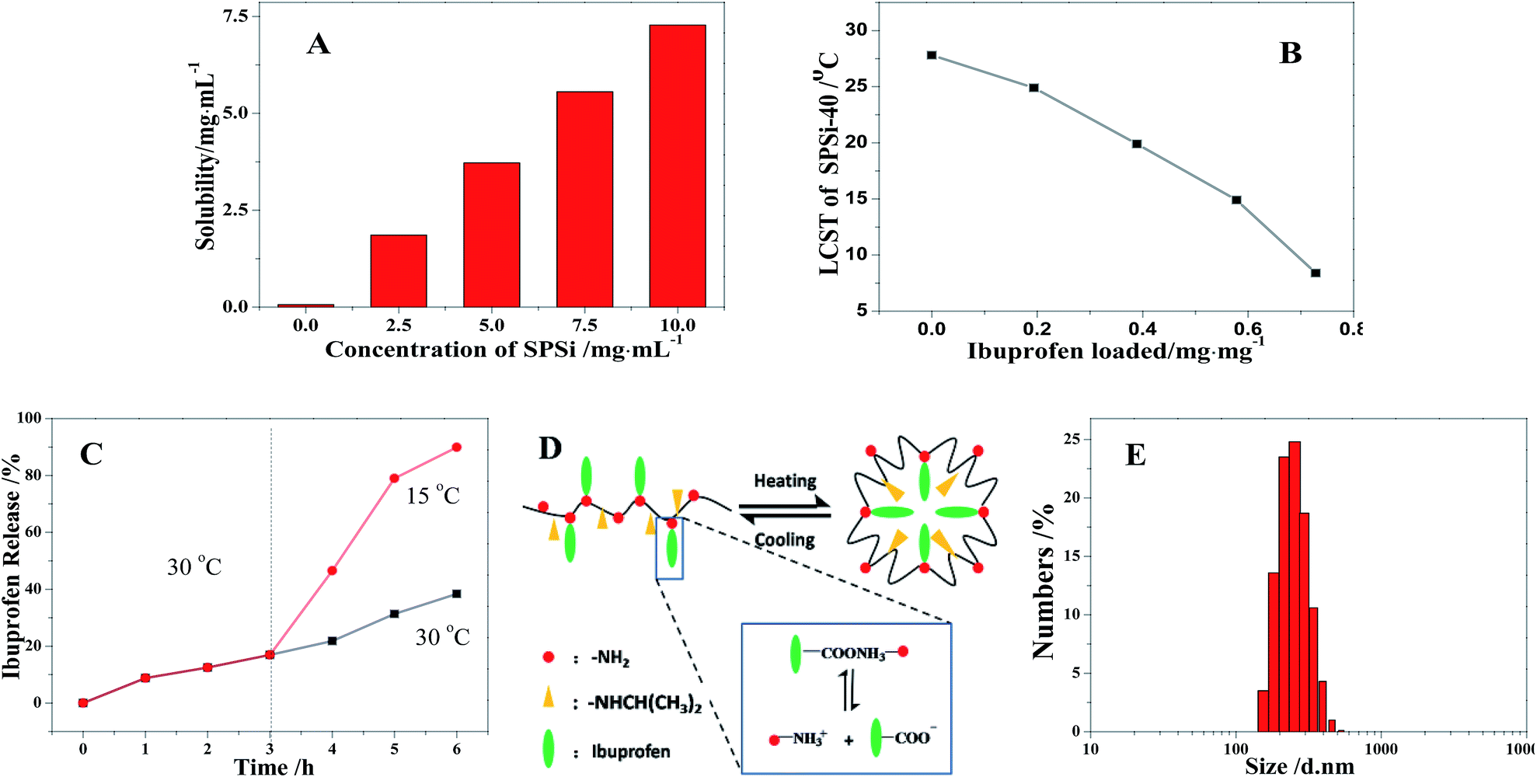
Various polymers have been widely studied to conjugate ibuprofen onto polymer backbone to achieve high drug loading and long-term release, as well as avoid the gastrointestinal side effects caused by the brush release of ibuprofen. As shown in Fig. 11A, the solubility of ibuprofen significantly increased from 0.06 mg mL−1 to 7.28 mg mL−1 as the concentration of the as-prepared SPSis-40 was increased from 0 mg mL−1 to 10 mg mL−1. The significant solubility should mainly originate from the intermolecular hydrogen bonding between the carboxyl of ibuprofen and amino group of SPSis. That is to say, high drug loading (72–74 wt%) is achieved, much more than ibuprofen encapsulated in polymer microparticles (less than 30 wt%) and covalent loaded onto polymers (40–60 wt%).52,53 When more ibuprofen (0 mg to 7.28 mg) was loaded onto SPSis-40 (10 mg mL−1), the LCST of SPSis-40 decreased gradually from 27.8 °C to 8.6 °C, because of the hydrophobic effect of loaded ibuprofen as pendant groups (Fig. 11B). At temperatures below the LCST, ibuprofen loaded SPSis precipitated from the solution and assembled into polymersomes. As shown in Fig. 11E, the assembled SPSis were of mean diameter of about 317 nm (PDI: 0.393) at pH 7.0 and 30 °C. Then, ibuprofen was encapsulated into the assembly. At high temperatures, ibuprofen was confined to the inner part and its release was restrained. When the temperature was reduced below the LCST, the SPSis-loaded drug became soluble into the aqueous solution again, and ibuprofen was rapidly released (Fig. 11C and D).
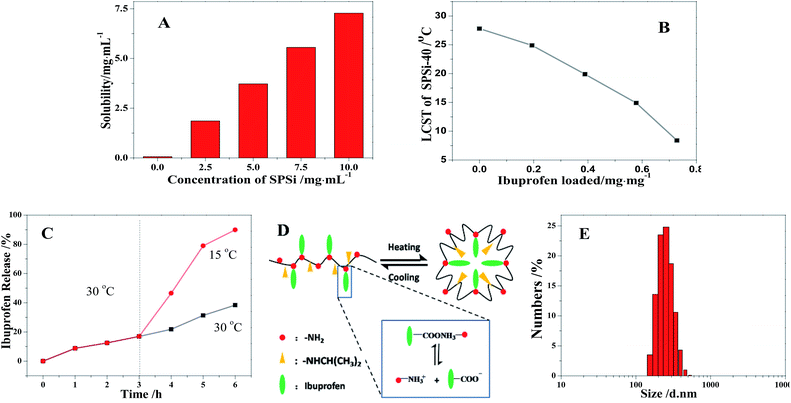 |
| | Fig. 11 Solubility of ibuprofen in SPSis-40 aqueous at varied concentration (A); LCST of SPSis-40 (10 mg mL−1) with different mass of ibuprofen loaded on (B); release profiles of ibuprofen loaded SPSis-40 (0.58 mg mg−1) at different temperature (C) and the schematic diagram of release mechanism (D); size of assembled SPSis-40 at pH 7.0 and 30 °C. | |
4. Conclusions
In summary, SPSis was successfully obtained via catalyst-free aza-Michael addition of NIPAM and PAPMS. The obtained SPSis exhibited phase transitions that were sensitive to temperature, pH, and salinity and phase separation occurred in 0.3 °C. By adjusting the molar ratio of amino/NIPAs, the LCST of SPSis could be varied over a broad range from 14.7 °C to 57.0 °C exponentially. The simulated relationship curve of NIPAs and LCST gives an accurate guide to obtain SPSis with predicted LCST. The LCST of SPSis decreased dramatically with increasing pH and salinity. The high sensitive of SPSis is due to its flexible Si–O–Si backbone. The obtained SPSis exhibited great potential of hydrophobic drug loading (about 75 wt%) as pendant groups by hydrogen bond and controlled release depended to temperature. Considering the efficient and facile synthesis of SPSis demonstrated in this study, preparing more advanced functional and smart polysiloxane systems for drug-delivery applications and biotherapy will be possible in the near future.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21274080) and Special Fund for Shandong Independent Innovation and Achievements Transformation (No. 2014ZZCX01101).
References
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- I. Tokarev and S. Minko, Adv. Mater., 2010, 22(31), 3446–3462 CrossRef CAS PubMed.
- B. A. Riggle, Y. Wang and I. J. Dmochowski, J. Am. Chem. Soc., 2015, 137(16), 5542–5548 CrossRef CAS PubMed.
- L. Dai, Q. Zhang, J. Li, X. Shen, C. Mu and K. Cai, ACS Appl. Mater. Interfaces, 2015, 7(13), 7357–7372 CAS.
- A. Higuchi, Q. D. Ling, S. S. Kumar, Y. Chang, T. C. Kao, M. A. Munusamy, A. A. Alarfaj, S. T. Hsu and A. Umezawa, Prog. Polym. Sci., 2014, 39, 1585–1613 CrossRef CAS.
- L. Xu, L. Feng, J. Hao and S. Dong, ACS Appl. Mater. Interfaces, 2015, 7, 8876–8885 CAS.
- N. Cakir, G. Hizal and C. R. Becer, Polym. Chem., 2015, 6, 6623–6631 RSC.
- G. K. Such, Y. Yan, A. P. Johnston, S. T. Gunawan and F. Caruso, Adv. Mater., 2015, 27, 2278–2297 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2012, 64, 49–60 CrossRef.
- I. Y. Konotop, I. R. Nasimova, M. V. Tamm, N. G. Rambidi and A. R. Khokhlov, Soft Matter, 2010, 6, 1632 RSC.
- H. Fu, X. Hong, A. Wan, J. D. Batteas and D. E. Bergbreiter, ACS Appl. Mater. Interfaces, 2010, 2(2), 452–458 CAS.
- H. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65(1), 10–16 CrossRef CAS PubMed.
- S. Peng and B. Bhushan, RSC Adv., 2012, 2, 8557–8578 RSC.
- A. Zintchenko, M. Ogris and E. Wagner, Bioconjugate Chem., 2006, 17, 766–772 CrossRef CAS PubMed.
- H. Chen, J. Li, Y. Ding, G. Zhang, Q. Zhang and C. Wu, Macromolecules, 2005, 38, 4403–4408 CrossRef CAS.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
- K. Ninomiya, S. Kawabata, H. Tashita and N. Shimizu, Ultrason. Sonochem., 2014, 21, 310–316 CrossRef CAS PubMed.
- M. Dou, D. C. Dominguez, X. Li, J. Sanchez and G. Scott, Anal. Chem., 2014, 86, 7978–7986 CrossRef CAS PubMed.
- G. B. Demirel, F. Buyukserin, M. A. Morris and G. Demirel, ACS Appl. Mater. Interfaces, 2012, 4, 280–285 CAS.
- J. R. Choi, Z. Liu, J. Hu, R. Tang, Y. Gong, S. Feng, H. Ren, T. Wen, H. Yang, Z. Qu, B. P. Murphy and F. Xu, Anal. Chem., 2016, 88(12), 6254–6264 CrossRef CAS PubMed.
- E. Yilgör and I. Yilgör, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef.
- J. B. Lin, B. C. Isenberg, Y. Shen, K. Schorsch, O. V. Sazonova and J. Y. Wong, Colloids Surf., B, 2012, 99, 108–115 CrossRef CAS PubMed.
- A. Car, P. Baumann, J. T. Duskey, M. Chami, N. Bruns and W. Meier, Biomacromolecules, 2014, 15, 3235–3245 CrossRef CAS PubMed.
- J. Tobis, Y. Thomann and J. C. Tiller, Polymer, 2010, 51, 35–45 CrossRef CAS.
- A. Mizutani, K. Nagase, A. Kikuchi, H. Kanazawa, Y. Akiyama, J. Kobayashi, M. Annaka and T. Okano, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2191–2198 CrossRef CAS PubMed.
- A. Mizutani, K. Nagase, A. Kikuchi, H. Kanazawa, Y. Akiyama, J. Kobayashi, M. Annaka and T. Okano, J. Chromatogr. A, 2010, 1217, 5978–5985 CrossRef CAS PubMed.
- C. Zhang, H. Peng, S. Puttick, J. Reid, S. Bernardi, D. J. Searles and A. K. Whittaker, Macromolecules, 2015, 48, 3310–3317 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- M. Igarashi, T. Matsumoto, T. Kobayashi, K. Sato, W. Ando, S. Shimada, M. Hara and H. Uchida, J. Organomet. Chem., 2014, 749, 421–427 CrossRef CAS.
- P. Gigler, M. Drees, K. Riener, B. Bechlars, W. A. Herrmann and F. E. Kühn, J. Catal., 2012, 295, 1–14 CrossRef CAS.
- H. Lu, L. Feng, S. Li, J. Zhang, H. Lu and S. Feng, Macromolecules, 2015, 48, 476–482 CrossRef CAS.
- L. W. Xu, J. W. Li, S. L. Zhou and C. G. Xia, New J. Chem., 2004, 28, 183–184 RSC.
- L. W. Xu and C. G. Xia, Synthesis, 2004, 2191–2195 CAS.
- L. W. Xu and C. G. Xia, Eur. J. Org. Chem., 2005, 4, 633–639 CrossRef.
- J. Škvarla, J. Zedník, M. Šlouf, S. Pispas and M. Štěpánek, Eur. Polym. J., 2014, 61, 124–132 CrossRef.
- C. Tonhauser, A. Alkan, M. Schömer, C. Dingels, S. Ritz, V. Mailänder, H. Frey and F. R. Wurm, Macromolecules, 2013, 46, 647–655 CrossRef CAS.
- M. Hruby, J. Kucka, O. Lebeda, H. Mackova, M. Babic, C. Konak, M. Studenovsky, A. Sikora, J. Kozempel and K. Ulbrich, J. Controlled Release, 2007, 119, 25–33 CrossRef CAS PubMed.
- O. Sergeeva, P. S. Vlasov, N. S. Domnina, A. Bogomolova, P. V. Konarev, D. I. Svergun, Z. Walterova, J. Horsky, P. Stepanek and S. K. Filippov, RSC Adv., 2014, 4, 41763–41771 RSC.
- K. Yoshimatsu, B. K. Lesel, Y. Yonamine, J. M. Beierle, Y. Hoshino and K. J. Shea, Angew. Chem., Int. Ed., 2012, 51, 2405–2408 CrossRef CAS PubMed.
- G. Fundueanu and M. Constantin, Acta Biomater., 2009, 5(1), 363–373 CrossRef CAS PubMed.
- Y. Y. Durmaz, Y. L. Lin and M. E. H. Elsayed, Adv. Funct. Mater., 2013, 23(31), 3885–3895 CrossRef CAS.
- S. Kumar and P. De, Polymer, 2014, 55, 824–832 CrossRef CAS.
- X. Yuan, B. Ju and S. Zhang, Carbohydr. Polym., 2014, 114, 530–536 CrossRef CAS PubMed.
- V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, Springer, 2011, pp. 29–89 Search PubMed.
- Č. Koňák, J. Pánek and M. Hrubý, Colloid Polym. Sci., 2007, 285, 1433–1439 Search PubMed.
- W. Jeong and Y. Lim, Bioconjugate Chem., 2014, 25, 1996–2003 CrossRef CAS PubMed.
- C. Luo, Y. Liu and Z. Li, Macromolecules, 2010, 43, 8101–8108 CrossRef CAS.
- I. M. Henderson, P. G. Adams, G. A. Montaño and W. F. Paxton, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 507–516 CrossRef CAS.
- S. Bayati, K. Zhu, L. T. Trinh, A. L. Kjoniksen and B. Nystrom, J. Phys. Chem. B, 2012, 116, 11386–11395 CrossRef CAS PubMed.
- P. C. H. Wong, P. W. S. Heng and L. W. Chan, Mol. Pharmaceutics, 2015, 12, 1592–1604 CrossRef CAS PubMed.
- R. R. Meléndez, W. Yu and K. E. Uhrich, Biomacromolecules, 2013, 14, 3542–3548 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.