
Synergy of graphene oxide–silver nanocomposite and amphiphilic co-polymer F127 on antibacterial properties and permeability of PVDF membrane†
Abstract
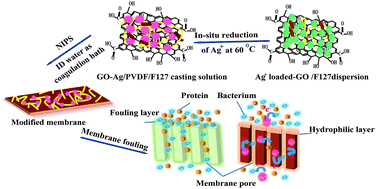
A graphene oxide–silver (GO–Ag) nanocomposite was formed by in situ reduction in the casting solution and was employed to decorate polyvinylidene fluoride (PVDF) membrane, together with amphiphilic co-polymer F127. The finger-like macro-voids morphology and surface roughness of modified membranes were presented by SEM and AFM microscopy. The FTIR spectrum and XPS analysis confirmed that the hydrophilic component contributed to the membrane wettability and permeability, while the graphene oxide–silver nanocomposite was conductive to membrane antimicrobial activity. Results indicated that under the comprehensive optimal conditions the prepared membranes possessed pure water flux of 269 L m−2 h−1 and BSA rejection ratio of 98.2%, the dynamic contact angles ranged from 60.5° to 10° within 80 s and the inhibition zone sizes against E. coli and S. aureus were 3 mm and 2 mm, respectively. Overall, the permeability, hydrophilicity and antibiofouling of modified membranes were regulated by the synergism between multifunctional modifier GO–Ag and F127 in a controllable blending-reaction modification process.
 Please wait while we load your content...
Please wait while we load your content...

