
Effect of lead and caesium on the mechanical, vibrational and thermodynamic properties of hexagonal fluorocarbonates: a comparative first principles study
 bc
and
bc
and
Abstract
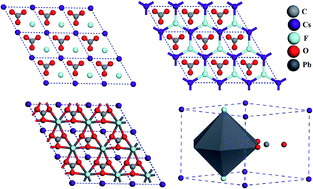
Exploration of the structure–property correlation of fluorocarbon materials has received much interest over recent years due to their extremely strong nonlinear optical (NLO) responses (13.6 times that of KH2PO4(KDP)), good ultraviolet (UV) cutoff (<200 nm) with better mechanical and chemical stability. In the present work a novel CsPbCO3F, ABCO3F (A = K, Rb; B = Ca, Sr) series is explored using density functional theory (DFT) calculations focusing on their mechanical, vibrational and thermodynamic properties and their Born effective charge (BEC) tensors. The calculated structural properties of lead carbonate fluoride with a semi-empirical dispersion corrected Ortmann Bechstedt Schmidt (OBS) method are found to be in relatively close agreement with experimental data. The obtained single crystal elastic constants satisfy the Born's mechanical stability criteria. The calculated bulk modulus value of lead carbonate (41 GPa) indicates its soft nature compared with other studied carbonates and is observed to be harder than KDP (26 GPa). In addition we have calculated the polycrystalline properties, bulk modulus (B), shear modulus (G), Young's modulus (E) and Poisson's ratio (σ) of CsPbCO3F and ABCO3F (A = K, Rb; B = Ca, Sr) using the Voigt, Reuss and Hill approximations. The obtained B/G (>1.75) results reveal the ductile nature of all the studied materials except for KCaCO3F (1.67) which is found to be brittle. Results of the hexagonal shear anisotropic factors (A1, A2, A3) indicate that all the studied crystals possess considerable mechanical anisotropy. Calculated zone centered vibrational infrared (IR) spectra confirm the higher optical activity of CsPbCO3F compared with the other carbonates. The obtained high frequency modes are consistent with the experimental values. The obtained BECs reveal the presence of a mixed covalent–ionic character of the compounds. The thermodynamic properties, namely entropy, Debye temperature, heat capacity, enthalpy, thermal expansion and thermal conductivity, have been computed at different temperatures ranging from 5 K to 1000 K. The results show that the lead based compound has the highest thermal conductivity (32.430 W m−1 K−1) of the reported carbonate materials. The results clearly indicate that the material could show better durability than LiNbO3, α-SiO2, CaCO3, and Ba3B6O12 hexagonal NLO materials. All the computed thermodynamic properties indicate that CsPbCO3F might be a potential candidate for second-order NLO applications. The polycrystalline, vibrational and thermodynamic properties of carbonate materials presented in this work could be a step forward in the process of developing new NLO materials.
 Please wait while we load your content...
Please wait while we load your content...

