DOI:
10.1039/C6RA20377A
(Paper)
RSC Adv., 2016,
6, 110422-110432
Luminescence properties of a Zn(II) supramolecular framework: easily tunable optical properties by variation of the alkyl substitution of (E)-N-(pyridine-2-ylethylidyne)arylamine ligands†
Received
12th August 2016
, Accepted 14th November 2016
First published on 15th November 2016
Abstract
A series of different alkyl substituted of Zn(II) complexes, [ZnL1Cl2] (Zn1), [ZnL2Cl2] (Zn2), [ZnL3Cl2] (Zn3), [ZnL4Cl2] (Zn4), [ZnL5Cl2] (Zn5) ((E)-N-(pyridine-2-yl)(CMe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPhR), where R = H, L1; 2-CH3, L2; 2,6-(CH3)2, L3; 2,4,6-(CH3)3, L4; 2-OCH3, L5) have been synthesized and characterized by single crystal X-ray diffraction, 1H NMR, FT-IR, and EA. The X-ray diffraction analyses revealed that although are all constructed by C–H⋯Cl/π hydrogen bonds and π⋯π interactions, the dimensions of these supramolecular frameworks complexes Zn1–Zn5 are quite different. Complexes Zn1 and Zn5 feature 3D 5-connected {46·64} and {44·66} topology structures, respectively, while complexes Zn2 and Zn3 feature 2D supramolecular layer with {63} topology structures. Complex Zn4 exhibits two different one-dimensional helix-shaped chains. Obviously, these results show that steric hindrance has a great impact on the final structures of the supramolecular architectures. Based on these varied structures caused by different alkyl substitutions, the emission maximum wavelengths of complexes Zn1–Zn5 can be tuned in a large range of 400–514 nm. The λem shift in the red direction after the substitution of alkyl is attributed to the HOMO–LUMO energy gap of complexes being effectively decreased due to the electron-donating ability of alkyl. These results are confirmed by the density functional theory calculations.
NPhR), where R = H, L1; 2-CH3, L2; 2,6-(CH3)2, L3; 2,4,6-(CH3)3, L4; 2-OCH3, L5) have been synthesized and characterized by single crystal X-ray diffraction, 1H NMR, FT-IR, and EA. The X-ray diffraction analyses revealed that although are all constructed by C–H⋯Cl/π hydrogen bonds and π⋯π interactions, the dimensions of these supramolecular frameworks complexes Zn1–Zn5 are quite different. Complexes Zn1 and Zn5 feature 3D 5-connected {46·64} and {44·66} topology structures, respectively, while complexes Zn2 and Zn3 feature 2D supramolecular layer with {63} topology structures. Complex Zn4 exhibits two different one-dimensional helix-shaped chains. Obviously, these results show that steric hindrance has a great impact on the final structures of the supramolecular architectures. Based on these varied structures caused by different alkyl substitutions, the emission maximum wavelengths of complexes Zn1–Zn5 can be tuned in a large range of 400–514 nm. The λem shift in the red direction after the substitution of alkyl is attributed to the HOMO–LUMO energy gap of complexes being effectively decreased due to the electron-donating ability of alkyl. These results are confirmed by the density functional theory calculations.
Introduction
The concept of “crystal engineering”1,2 was revolutionized with the participation of coordination polymers and metallosupramolecular architectures.3–6 The latter is not only designed and developed for their diverse structural aesthetics but also for a variety of potential applications in catalysis, separation, sensors, adsorption, luminescent materials, magnetism, etc.7–11 The glue for the inorganic supramolecular synthesis is the metal–ligand coordinate-covalent bond whose strength and directionality have been thoroughly exploited for engineering various molecular building blocks into diverse supramolecular architectures.12–14 The dimensionality of the resulting coordination network can be increased by noncovalent interactions, such as hydrogen bonds, π⋯π, C–H⋯π, aromatic ring⋯halogen, and/or other van der Waals interactions.15–20 Schiff base ligands are frequently used in coordination chemistry due to their significant ability to form stable complexes with metalions.21,22 Zinc Schiff base complexes with luminescent properties can be used in applications involving fabrication of novel materials and as probes in biological systems.23 Controllable tuning of fluorescent properties of zinc complexes based on ligand design is always a challenge for chemists. From the viewpoint of molecular and crystal engineering, the critical criterion for ligand design is how to embed noncovalent interaction groups to control the molecular structure and the supramolecular framework.
The type and position of substituents are the main factors to affect the supramolecular frameworks and luminescent properties. In line with the above discussion, five (E)-N-(pyridine-2-ylethylidyne)arylamine ligands, L1–L5, Scheme 1, carrying different alkyl substitutions in the phenyl ring have been employed for the synthesis of Zn(II) complexes. Five Zn(II) complexes, namely Zn1–Zn5, were prepared by the reactions of corresponding ligand and zinc chloride, which display luminescence ranging from deep blue to bluish green. The optical properties of all the complexes were investigated by UV-vis absorption and luminescence spectroscopy in solution and in the solid state. The electronic effects of the substituents on the ligand were also determined as important factors for the modulation of the luminescence wavelength. In this report we will also carry out density functional theory (DFT) calculations in both the ground and excited state of the complexes for understanding of the electronic structures.
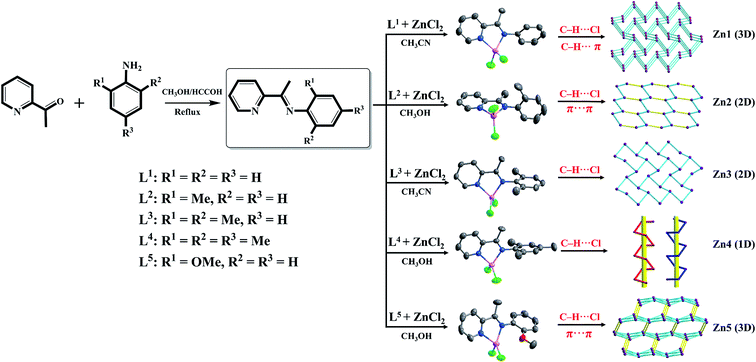 |
| | Scheme 1 Syntheses routes of five ligands L1–L5 and the corresponding Zn(II) complexes. | |
Experimental section
Materials and instrumentation
All reagents and solvents were purchased from commercial sources and were used without further purification. Elemental analyses were carried out on a Perkin-Elmer 2400 automatic analyzer. FT-IR spectra data (4000–400 cm−1) were collected by a Nicolet impact 410 FT-IR spectrometer. 1H NMR spectra were obtained using a Bruker Avance-400 MHz spectrometer with Si(CH3)4 as internal standard. The thermal analyses were performed on a ZRY-2P thermogravimetric analysis under a flow of air from room temperature to 700 °C. Powder X-ray diffraction (PXRD) patterns were recorded in the 2θ range of 5–50° using Cu-Kα radiation by Shimadzu XRD-6000 X-ray Diffractometer. A Perkin-Elmer Lambda 35 spectrometer was used to measure the UV-vis absorption spectra of ligands and complexes. The emission luminescence and lifetime properties were recorded with Edinburgh FLS 920 fluorescence spectrometer.
X-ray crystallography
Suitable crystals of complexes Zn1–Zn5 were selected and mounted on a Rigaku R-AXIS RAPID IP diffractometer. Diffraction data were collected using graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å). The structures were solved with direct methods24 and refined with full-matrix least-squares on F2. All the hydrogen atoms were constrained in geometric positions to their parent atoms and non-hydrogen atoms were refined anisotropically. The detailed crystal structure refinement data are given in Table 1, selected bond lengths and angles are listed in Tables S1 and S2.† The CCDC numbers are 1494305–1494308, 1477759 for Zn1–Zn5, respectively.†
Table 1 Crystallographic and structural determination data for complexes Zn1–Zn5
| |
Zn1 |
Zn2 |
Zn3 |
Zn4 |
Zn5 |
| R1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. |
| CCDC no. |
1494305 |
1494306 |
1494307 |
1494308 |
1477759 |
| Formula |
C13H12Cl2N2Zn |
C14H14Cl2N2Zn |
C15H16Cl2N2Zn |
C16H18Cl2N2Zn |
C14H14Cl2N2OZn |
| Mr |
332.52 |
346.56 |
360.57 |
374.61 |
362.54 |
| Cryst. syst. |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
C2/c |
P21/n |
| a [Å] |
9.558(5) |
8.108(5) |
9.095(4) |
32.804(14) |
11.311(3) |
| b [Å] |
10.911(5) |
13.089(8) |
11.742(5) |
13.390(5) |
9.796(3) |
| c [Å] |
13.640(6) |
15.690(9) |
15.094(7) |
16.187(6) |
14.305(4) |
| α [°] |
90 |
71.786(7) |
90 |
90 |
90 |
| β [°] |
92.856(4) |
76.105(7) |
91.716(10) |
96.815(8) |
106.827(3) |
| γ [°] |
90 |
84.537(7) |
90 |
90 |
90 |
| Volume [Å3] |
1420.82(12) |
1535.1(16) |
1611.48(12) |
7060(5) |
1517.1(8) |
| Z |
4 |
4 |
4 |
16 |
4 |
| Dc [g cm−3] |
1.554 |
1.499 |
1.486 |
1.410 |
1.587 |
| μ [mm−1] |
2.087 |
1.935 |
1.846 |
1.689 |
1.966 |
| F(000) |
672 |
704 |
736 |
3071 |
736 |
| θ range [°] |
3.25–27.56 |
1.40–26.81 |
3.11–27.58 |
1.25–25.00 |
2.03–26.93 |
| h range |
−12 ≤ h ≤ 12 |
−10 ≤ h ≤ 10 |
−11 ≤ h ≤ 11 |
−38 ≤ h ≤ 38 |
−13 ≤ h ≤ 14 |
| k range |
−12 ≤ k ≤ 14 |
−16 ≤ k ≤ 15 |
−15 ≤ k ≤ 15 |
−15 ≤ k ≤ 15 |
−12 ≤ k ≤ 12 |
| l range |
−6 ≤ l ≤ 17 |
−19 ≤ l ≤ 15 |
−19 ≤ l ≤ 19 |
−19 ≤ l ≤ 19 |
−18 ≤ l ≤ 17 |
| Data/restraints/params |
3241/0/163 |
5771/30/346 |
3722/0/181 |
6209/0/379 |
3041/0/181 |
| GOF |
0.808 |
1.034 |
1.003 |
1.063 |
1.023 |
| R1, wR2[I > 2σ(I)]a |
0.0390, 0.1119 |
0.0382, 0.0944 |
0.0405, 0.1224 |
0.0405, 0.1454 |
0.0336, 0.0807 |
| R1, wR2[all data]a |
0.0583, 0.1309 |
0.0681, 0.1084 |
0.0541, 0.1350 |
0.0515, 0.1580 |
0.0479, 0.0873 |
| Δρmax, Δρmin [e Å−3] |
0.346, −0.418 |
0.448, −0.530 |
0.601, −0.341 |
1.435, −0.997 |
0.379, −0.370 |
Computational details
To gain insights into the photophysical properties of complexes Zn1–Zn5, and the energy transfer process from the free ligand to the complexes, their energy levels based on density functional theory (DFT) theoretical calculations were performed. The geometries obtained from X-ray diffraction analyses were fully optimized first in conjunction with the acetonitrile solvent model based on B3LYP/6-31G basis set. The theoretical investigation of geometry optimization was performed with the Gaussian 09 program package.25 Density functional theory (DFT) was calculated at Beck's three-parameter hybrid exchange functional26 and Lee, and Yang and Parr correlation functional27 B3LYP/6-31G(d). The spin density distributions were visualized using Gaussview 5.0.8.
General synthesis of zinc(II) complexes
The zinc(II) complexes were synthesized by dissolving metal salts ZnCl2 and 1 molar equivalence of the respective Schiff-base ligands in anhydrous solutions (acetonitrile or methanol). The yellow solutions were refluxed for 12 h and subsequently filtered. X-ray quality single crystals of five Zn(II) complexes were grown from slow evaporation of their solutions (Scheme 1). In addition, the detailed IR and 1H NMR data for complexes Zn1–Zn5 are listed in the Table S3.†
[Zn(L1)Cl2] (Zn1). Yield: 43.2 mg (65%). Anal. calcd (%) for C13H12Cl2N2Zn (M = 332.52 g mol−1): C, 46.96; H, 3.64; N, 8.42. Found: C, 46.92; H, 3.71; N, 8.45. FT-IR bands (KBr, cm−1): νCN 1623, νZn–N 446.
[Zn(L2)Cl2] (Zn2). Yield: 40.3 mg (58%). Anal. calcd (%) for C14H14Cl2N2Zn (M = 346.56 g mol−1): C, 48.52; H, 4.07; N, 8.08. Found: C, 48.55; H, 4.14; N, 8.03. FT-IR bands (KBr, cm−1): νCN 1632, νZn–N 438.
[Zn(L3)Cl2] (Zn3). Yield: 38.2 mg (53%). Anal. calcd (%) for C15H16Cl2N2Zn (M = 360.57 g mol−1): C, 49.96; H, 4.47; N, 7.77. Found: C, 50.03; H, 4.48; N, 7.66. FT-IR bands (KBr, cm−1): νCN 1631, νZn–N 439.
[Zn(L4)Cl2] (Zn4). Yield: 43.5 mg (58%). Anal. calcd (%) for C16H18Cl2N2Zn (M = 374.61 g mol−1): C, 51.30; H, 4.84; N, 7.48. Found: C, 51.33; H, 4.89; N, 7.52. FT-IR bands (KBr, cm−1): νCN 1623, νZn–N 433.
[Zn(L5)Cl2] (Zn5). Yield: 47.4 mg (65%). Anal. calcd (%) for C14H14Cl2N2OZn (M = 362.54 g mol−1): C, 46.38; H, 3.89; N, 7.73. Found: C, 46.41; H, 3.95; N, 7.70. FT-IR bands (KBr, cm−1): νCN 1620, νZn–N 437.
Results and discussion
Synthesis and spectral characterization
The five imine ligands (E)-N-(pyridine-2-yl)(CMeNPhR) (where R = H, L1; 2-CH3, L2; 2,6-(CH3)2, L3; 2,4,6-(CH3)3, L4; 2-OCH3, L5) were readily prepared in a one-step procedure by condensation of 2-acetylpyridine with the corresponding monamine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in anhydrous methanol/formic acid solution (see Scheme 1). Yields for the five imine ligands varied from 81 to 92%. Using the self-assembled method, these ligands were reacted with ZnCl2 to obtain a series of complexes, namely, Zn1–Zn5. X-ray quality single crystals of five Zn(II) complexes were grown from slow evaporation of methanol or acetonitrile solutions and readily obtained in good yields within the range of 53–65%. The details of the synthesis are given in the Experimental section. We confirm that all the complexes are stable in the solid state under exposure to air. The complexes are soluble in common organic solvents, such as chloroform, dimethylsulfoxide and acetonitrile.
:1 molar ratio in anhydrous methanol/formic acid solution (see Scheme 1). Yields for the five imine ligands varied from 81 to 92%. Using the self-assembled method, these ligands were reacted with ZnCl2 to obtain a series of complexes, namely, Zn1–Zn5. X-ray quality single crystals of five Zn(II) complexes were grown from slow evaporation of methanol or acetonitrile solutions and readily obtained in good yields within the range of 53–65%. The details of the synthesis are given in the Experimental section. We confirm that all the complexes are stable in the solid state under exposure to air. The complexes are soluble in common organic solvents, such as chloroform, dimethylsulfoxide and acetonitrile.
The infrared spectra of these five Zn(II) complexes (see Fig. S1 and S2†) are similar to that of the corresponding ligand and the IR assignments of selected diagnostic bands are given in the Experimental section. In the IR spectra of ligands L1–L5, the existence of CN bonds is clearly demonstrated by the presence of strong characteristic CN peaks in a range of 1625–1657 cm−1. These bands undergo negative shifts of 2–34 cm−1 in the complexes, which may be attributed to the coordination of the nitrogen atom of the imine to the metal ion.28,29 This is further confirmed by the presence of a ν(M − N) vibration in the region 433–446 cm−1 in all the complexes.30 The 1H NMR spectra of the imine ligands L1–L5 and the corresponding Zn(II) complexes were recorded in CDCl3 at room temperature (see Fig. S3 and S4†). In the 1H NMR spectra, the chemical shift values of the protons in the complexes are slightly different from those observed for the non-coordinated ligand. Of particular note is the Py-H6 resonances of the complexes Zn1–Zn5, which are moved down-field due to coordination between the pyridine nitrogen and the zinc(II) center. Meanwhile, the resonance peaks at 3.87 (L5) and 4.08 (Zn5) ppm are assigned to –OCH3 protons.
The thermal stability of the five complexes was investigated by the TGA technique (see Fig. S5, ESI†). The TGA curves of complexes Zn1–Zn3 are similar and thermally decomposed in two steps. The first weight loss of 20.63% at around 271 °C, due to the removal of 2HCl molecules (calc. 21.32%) in Zn1, and 25.22% at around 291 °C, due to the removal of 2HCl + (–CH3) molecules (calc. 24.78%) in Zn2, and 27.70% at around 310 °C, due to the removal of 2HCl + 2(–CH3) molecules (calc. 27.98%) in Zn3, respectively. The following decompositions of Zn1–Zn3 in the temperature range of 381–649 °C. For Zn4 and Zn5, there are three continuous weight loss steps among them. The first weight loss can be attributed 2HCl molecules, which is approximately 19.44% for Zn4 and 19.13% for Zn5 (calc. 18.92% for Zn4 and 19.55% for Zn5). The following weight losses of 11.03% for Zn4 are attributed to the loss of three –CH3 (calc. 12.01%) and 14.21% for Zn5 can ascribe to the release of one –OCH3 and one –CH3 (calc. 12.69%). These complexes finally transformed to the final residual of the ZnO phase (calc. 24.47%, 23.48%, 22.57%, 18.92%, and 22.44% for Zn1–Zn5, respectively), which are in agreement with the experimental results (found 24.97%, 23.54%, 22.81%, 19.13%, and 22.81% for Zn1–Zn5, respectively).
We performed powder X-ray diffraction patterns on complexes Zn1–Zn5 to check the purity of the bulk products. From Fig. S6,† we can see that all major peak positions of the measured patterns are in good agreement with those simulated. Furthermore, the differences in intensity may be due to the preferred orientation of the crystal products. These observations are consistent with the crystal structure (see below). To further understand the structures of these complexes, single crystals were obtained and analyzed by single crystal X-ray diffraction. The crystallographic data for these complexes Zn1–Zn5 are listed in Table 1.
Description of the structures
[Zn(L1)Cl2] (Zn1). Complex Zn1 crystallized in the monoclinic space group P21/c. As shown in Fig. 1a, the asymmetric unit of Zn1 consists of one Zn(II) atom, one L1 ligand and two chlorine anions. Using Addison's model,31,32 the coordination geometry around the zinc atom in Zn1 (τ4 = 0.890) can be better described as a tetrahedron. It is noteworthy that it gives rise to a five-membered ring after coordinated featuring coplanarity with inherency of pyridyl ring and indirectly results the extended conjugate. In complex Zn1, the pyridine ring and the phenyl rings of L1 ligand are twisty and the dihedral angle is ∼60.441°. The average bond lengths of Zn–N and Zn–Cl are 2.050(2) and 2.193(5) Å, respectively. The bond angles around the Zn(II) ion are in the range of 79.78(1)–118.26(8)°. The selected bond lengths and bond angles of complex Zn1 are given in Table S1.† Single crystal analysis shows that the H⋯Cl distances (2.678 Å and 2.896 Å) between the CH and the chlorine atoms from neighboring molecules is shorter than the sum of the van der Waals radii for H and Cl (ca. 1.2 Å for H, 1.75 Å for Cl),33 and the C–H⋯Cl angles are 130.782° and 130.422°, which indicates a satisfactory intermolecular hydrogen bonds. First, the C7–H7C⋯Cl2 hydrogen bonds from acetyl lead to generation of the dimers (the distance of H7C⋯Cl2 is 2.678 Å), which is shown in Fig. 1b. Then the C12–H12A⋯Cl1 hydrogen bonds linked these dimers to generate a two-dimensional layer (the distance of H12A⋯Cl1 is 2.896 Å). The two-dimensional layer further extend into three-dimensional supramolecular network through C3–H3A⋯π interactions (the distance of H3A⋯π is 3.036 Å), as shown in Fig. 1c. The detailed data of C–H⋯Cl hydrogen bonds and C–H⋯π interactions for Zn1 are listed in Table 2. If the mononuclear unit is viewed as the nodes and the C–H⋯Cl and C–H⋯π interactions as the aqua and red linkers, respectively, the resulting 5-connected three-dimensional supramolecular structure may be simplified as a bnn net with the Schläfli symbol of {46·64}, as depicted in Fig. 1c.
 |
| | Fig. 1 (a) Crystal structure of Zn1. Thermal ellipsoid is drawn at 50% probability. (b) The 2D layer structure in Zn1. (c) The 3D network structure and topological view in Zn1. Dotted lines represent the C–H⋯Cl and C–H⋯π interactions. | |
Table 2 Structural and geometrical parameters for complexes Zn1–Zn5
| Complex |
Addison parameter (τ4) |
Dihedral anglea (degree) |
D–H⋯A |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
D–H⋯A (degree) |
Supramolecular dimension |
| Between the pyridyl ring and phenyl ring. Cg1 = phenyl ring. Cg2 = pyridyl ring. |
| Zn1 |
0.890 |
60.441 |
C7–H7C⋯Cl2 |
0.960 |
2.678 |
3.384 |
130.782(2) |
3D |
| C12–H12A⋯Cl1 |
0.930 |
2.896 |
3.570 |
130.422(2) |
| C3–H3A⋯πCg1b |
|
|
3.036 |
|
| Zn2 |
0.899, 0.891 |
83.403, 85.146 |
C2–H2A⋯Cl4 |
0.930 |
2.889 |
3.563 |
133.430(4) |
2D |
| C3–H3A⋯Cl1 |
0.930 |
2.897 |
3.702 |
145.695(5) |
| C17–H17A⋯Cl3 |
0.930 |
2.890 |
3.685 |
144.210(5) |
| πCg1⋯πCg1 |
|
|
3.741, 3.851 |
|
| Zn3 |
0.876 |
80.205 |
C4–H4A⋯Cl1 |
0.930 |
2.908 |
3.561 |
128.428(2) |
2D |
| C7–H7A⋯Cl1 |
0.960 |
2.916 |
3.656 |
134.690(2) |
| Zn4 |
0.901, 0.879 |
87.987, 84.936 |
C3–H3A⋯Cl3 |
0.930 |
2.895 |
3.735 |
150.869(2) |
1D |
| C19–H19A⋯Cl1 |
0.930 |
2.934 |
3.716 |
142.632(2) |
| C30–H30B⋯Cl2 |
0.960 |
2.889 |
3.689 |
141.490(3) |
| Zn5 |
0.791 |
49.456 |
C4–H4A⋯Cl2 |
0.930 |
2.741 |
3.648 |
165.258(2) |
3D |
| C10–H10A⋯Cl2 |
0.930 |
2.875 |
3.746 |
156.511(1) |
| πCg2⋯πCg2c |
|
|
|
|
[Zn(L2)Cl2] (Zn2). Crystal refinement data of complex Zn2 implied that it belongs to a triclinic system, P space group. The asymmetric unit consists of two crystallographically and conformationally independent molecules as shown in Fig. 2a. In Zn2, two zinc atoms are bonded to four nitrogen atoms with similar distances varying from 2.055(3) to 2.073(3) Å (Table S2†). The coordination geometry of Zn2+ are distorted tetrahedron, with four-coordinated geometry index τ4 = 0.899 (Zn1) and 0.891 (Zn2). The bond angles around the Zn(II) ion are in the range of 79.32(1)–118.15(9)°. The dihedral angles between the pyridyl ring and the phenyl ring in the two molecules are 83.403° and 85.146°, respectively (Table 2), which are seriously twisty compared with the complex Zn1. From Fig. 2b, the independent units are linked through C2–H2A⋯Cl4, C3–H3A⋯Cl1 and C17–H17A⋯Cl3 hydrogen bonds interactions to generate a one-dimensional “wave-like” chain. The H2A⋯Cl4, H3A⋯Cl1 and H17A⋯Cl3 distances are 2.859, 2.890 and 2.897 Å, respectively. In Zn2, except the C–H⋯Cl hydrogen bonds interactions, the intermolecular π⋯π stacking interactions play important roles in assembling the two-dimensional structures. As shown in Fig. 2c, a 2D layer can be constructed by interlayer πphenyl⋯πphenyl (3.741 and 3.851 Å) stacking interactions. Topology analysis shows that the overall network of Zn2 features a 2D hcb topology structure with a Schläfli symbol of {63} by denoting the mononuclear units as 3-connected nodes and the C–H⋯Cl and π⋯π interactions as the aqua and yellow linkers, respectively (Fig. 2d).
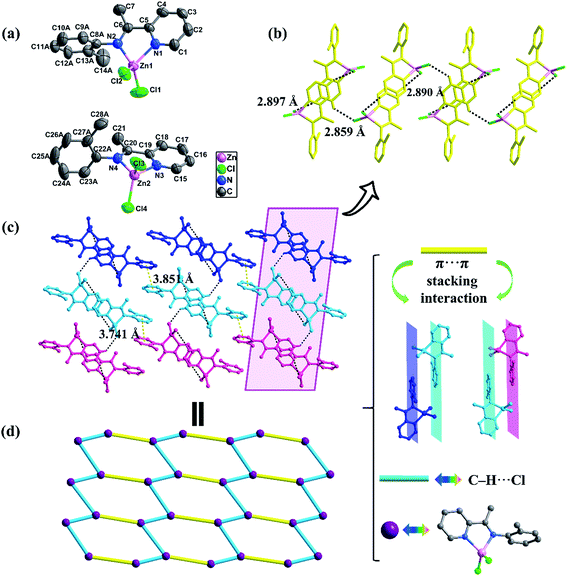 |
| | Fig. 2 (a) Crystal structure of Zn2. Thermal ellipsoid is drawn at 50% probability. H atoms have been omitted for clarity. (b) The 1D “wave-like” chain structure in Zn2. (c) The 2D layer structure in Zn2. (d) Topological representation of the 2D layer structure in Zn2. Dotted lines represent the C–H⋯Cl and π⋯π interactions. | |
[Zn(L3)Cl2] (Zn3). Complex Zn3 belongs to the monoclinic P21/n space group. In complex Zn3, the Zn(II) center adopts a distorted tetrahedral geometry (τ4 = 0.876) coordinated by two nitrogen atoms of (E)-2,6-dimethyl-N-((pyridin-2-yl)ethylidene)aniline (L3) and two terminal chlorine ions (Fig. 3a). In particular, the Zn(II) cation coordinates with nitrogen atoms from ligand L3 to form a five membered ring, which further extends the conjugated system. The dihedral angle between the pyridyl ring and the phenyl ring is 80.205° in complex Zn3 (Table 2), which indicates that the ligand L3 displays enormously twisty in Zn3. Likewise, the distances of Zn–N (imine) are relatively shorter than that of Zn–N (pyridine). The average distances of the Zn–N bonds are 2.066(3) Å. The selected bond lengths and bond angles of complex Zn3 are given in Table S1.† In the complex Zn3, the intermolecular C7A–H7A⋯Cl1 hydrogen bonds interactions with H7A⋯Cl1 distance of 2.916 Å to bridge two mononuclear units to generate one-dimensional “wave-like” chains, as shown in Fig. 3b. Meanwhile, the C4–H4A⋯Cl1 hydrogen bonds from pyridine lead to generation of the dimers (the distance of H4A⋯Cl1 is 2.908 Å). Through these weak interactions a two-dimensional layer is constructed, as shown in Fig. 3b, in which each molecule links three adjacent ones. In order to predict the structure, we view the mononuclear unit as a 3-connected node and the C–H⋯Cl hydrogen bonds interactions as the aqua linker, as shown in Fig. 3c. Thus, the overall topology of Zn3 can be defined as the {63} hcb topology.
 |
| | Fig. 3 (a) Crystal structure of Zn3. Thermal ellipsoid is drawn at 50% probability. (b) The 2D layer structure in Zn3. (c) Topological representation of the 2D layer structure in Zn3. Dotted lines represent the C–H⋯Cl interactions. | |
[Zn(L4)Cl2] (Zn4). Similar to complex Zn2, complex Zn4 also displays two crystallographically and conformationally independent molecules (Fig. S7†) of identical composition in the crystal but with different behaviors. The differences of lengths and angles between center ion and coordinated atoms are tiny (see Table S2†). In complex Zn4, the zinc(II) ions are four-coordinated, and the coordination geometry of Zn2+ are distorted tetrahedron, with geometry index τ4 = 0.901 (Zn1) and 0.879 (Zn2). Due to the larger steric hindrance of 2,4,6-(CH3)3 substitution, the pyridyl and phenyl rings of the L4 ligand in Zn4 are arranged in a nearly perpendicular fashion (87.987°/84.936°). The bond angles around the Zn(II) ion are in the range of 79.69(1)–119.01(8)°. Meanwhile, the steric arrangements of them in crystal are slightly different. The two mononuclear units are assembled through intermolecular hydrogen bonds C3–H3A⋯Cl3 of 2.895 Å, C19–H19A⋯Cl1 of 2.934 Å and C30–H30B⋯Cl2 of 2.889 Å respectively, to form two different one-dimensional chains, as shown in Fig. 4a. If the mononuclear unit is viewed as the nodes and the C–H⋯Cl hydrogen bonds interactions as the red and blue linkers, the resulting one-dimensional supramolecular structure may be simplified as the right- and left-handed helix chains, as shown in Fig. 4b.
 |
| | Fig. 4 (a) The 1D chain structures in Zn4. (b) Representation of the right- and left-handed helix chains in Zn4. Dotted lines represent the C–H⋯Cl interactions. | |
[Zn(L5)Cl2] (Zn5). The single-crystal X-ray diffraction analysis reveals that complex Zn5 crystallizes in the monoclinic system, space group C2/c with one molecule in the unit cell (Fig. 5a). As shown in Fig. 5b, the coordination number of Zn(II) is four (τ4 = 0.791) with two nitrogen atoms from ligand L5 and two terminal chlorine atoms. Complex Zn5 has the dihedral angle of 49.456°, between pyridyl ring and phenyl ring (Fig. 5c), which indicates that Zn5 displays better coplanarity than Zn1–Zn4. The distances of Zn1–N1 and Zn1–N2 are 2.087(2) Å and 2.083(2) Å, in agreement with the values of the similar zinc complex.34 The detailed bond distances and angles are listed in Table S1.† In complex Zn5, the independent units are linked through hydrogen bonds C4–H4A⋯Cl2 of 2.741 Å and C10–H10A⋯Cl2 of 2.875 Å to generate a two-dimensional supramolecular layer (Fig. 5d). In addition to the intermolecular hydrogen bonds C–H⋯Cl, π⋯π stacking plays an important role in assembling the mononuclear molecule [Zn(L5)Cl2] into a three-dimensional supramolecular network (Fig. 5e). From Fig. 5e, these 2D sheets are further assembled into a 3D supramolecular network by inter-layer π⋯π interactions from the two pyridine rings (the shortest distance between the two pyridine rings is about 3.770 Å). The final supramolecular architecture can be simplified into a 3D 5-connected sqp topology with the Schläfli symbol of {44·66} (Fig. 5f) under the circumstance of the mononuclear unit viewed as the nodes and the C–H⋯Cl and π⋯π interactions as the aqua and yellow linkers, respectively.
 |
| | Fig. 5 (a) Crystal structure of Zn5. Thermal ellipsoid is drawn at 50% probability. (b) Coordination geometry of zinc(II) ions. (c) Depiction of the dihedral angle between the pyridine ring and phenyl ring in Zn5. (d) The 2D layer structure in Zn5. (e) The 3D network structure of and (f) topological view in Zn5. Dotted lines represent the C–H⋯Cl and π⋯π interactions. | |
Photophysical studies
Absorption properties of the zinc(II) complexes in CH3CN. The UV-vis absorption spectra of the ligands L1–L5 and complexes Zn1–Zn5 were recorded at room temperature in CH3CN (10 μmol L−1). Because of ligands with similar structure, the UV-vis spectra are similar (Fig. S8†). Fig. 6 shows the absorption spectra of complexes Zn1–Zn5 in acetonitrile exhibit two distinct absorption bands at ca. 280 nm and ca. 330 nm. The numerical values of the maximum absorption wavelength and molar extinction coefficients (ε) are listed in Table 3. For complexes Zn1–Zn5, these absorption bands are similar to the corresponding ligands which can be assigned as intraligand charge transfer transitions (ILCT)-type.35 The low energy absorption bands of complexes Zn2–Zn5 are 332, 337, 350, 354 nm, respectively, which shift to longer wavelengths compared to that of Zn1 (323 nm) due to the increase of electron-donating groups. Interestingly, the introductions of electron-donating (–CH3 and –OCH3) groups both result in a 9–31 nm bathochromic shift of the absorption bands, and these effects are cumulative, with an increase in the number of CH3.
 |
| | Fig. 6 UV-vis absorption spectra of Zn1–Zn5 in CH3CN at room temperature. | |
Table 3 Photophysical properties of the ligands L1–L5 and complexes Zn1–Zn5
| Complex |
Absorptiona |
Photoluminescence in acetonitrile |
Photoluminescence in the solid state |
| λabs/nm (ε/M−1 cm−1) |
λem/nm |
FWHM/nm |
τ/μs |
ΦPLb |
CIE (x, y) |
λem/nm |
FWHM/nm |
τ/μs |
CIE (x, y) |
| Measured in CH3CN solutions (∼1 × 10−5 M). Take quinine sulfate in CH3CN as reference (∼1 × 10−5 M, ΦPL = 0.546). |
| L1 |
269 (12895), 323 (1357) |
393 |
80.15 |
6.10 |
0.020 |
0.19, 0.13 |
458 |
147.11 |
7.93 |
0.18, 0.25 |
| Zn1 |
280 (40427), 323 (18193) |
400 |
101.40 |
9.15 |
0.095 |
0.17, 0.12 |
472 |
134.85 |
9.90 |
0.22, 0.29 |
| L2 |
269 (20688), 329 (2216) |
397 |
134.20 |
8.10 |
0.025 |
0.18, 0.11 |
465 |
177.55 |
8.33 |
0.21, 0.27 |
| Zn2 |
280 (42341), 332 (5882) |
412 |
69.14 |
8.54 |
0.160 |
0.16, 0.07 |
480 |
147.14 |
9.13 |
0.25, 0.34 |
| L3 |
269 (10543), 334 (1195) |
402 |
82.40 |
8.62 |
0.028 |
0.20, 0.19 |
470 |
96.04 |
8.73 |
0.25, 0.29 |
| Zn3 |
281 (42808), 337 (2579) |
416 |
72.44 |
8.66 |
0.193 |
0.16, 0.07 |
495 |
104.34 |
9.64 |
0.23, 0.37 |
| L4 |
268 (18246), 340 (1697) |
413 |
112.42 |
7.43 |
0.046 |
0.17, 0.13 |
500 |
155.62 |
7.55 |
0.30, 0.39 |
| Zn4 |
281 (45048), 350 (2829) |
436 |
105.03 |
8.08 |
0.362 |
0.17, 0.15 |
514 |
154.14 |
8.37 |
0.28, 0.38 |
| L5 |
270 (17357), 346 (2200) |
405 |
126.15 |
7.27 |
0.037 |
0.18, 0.13 |
480 |
152.64 |
8.91 |
0.29, 0.35 |
| Zn5 |
281 (36371), 354 (19852) |
422 |
65.15 |
8.04 |
0.278 |
0.16, 0.06 |
499 |
187.57 |
10.25 |
0.31, 0.38 |
Solid-state and solution luminescence properties of the zinc(II) complexes. Luminescent zinc(II) complexes possessing closed d10 shells are superior potential candidates as valuable luminescent materials;36–38 thus, we probed the luminescence properties of Zn1–Zn5 in the solid state and acetonitrile solution at room temperature. Selected data are summarized in Table 3. In the solid state, the free ligands L1–L5 exhibit bluish green emission bands centered at 458, 465, 470, 500, 480 nm (Fig. S9a and b†), respectively, which may be attributed to the π*–π transitions.39,40 For the complexes Zn1–Zn5, the emission bands are observed at 472, 480, 495, 514, and 499 nm (Fig. 7a and b), respectively. Due to the similarity of the emission bands with that of the corresponding ligand, the emissions of these zinc(II) complexes may be attributed to the intraligand transitions.41,42 In addition, the emission bands of Zn1–Zn5 occur red-shifted compared to that of the corresponding ligand, which may be attributed to the coordination of ligand to the metal centers. A trend is observed in λem with Zn1 < Zn2 < Zn3 < Zn5 < Zn4 which is consistent with the electron-donating ability (H < –CH3 < –OCH3) of substituents of these ligands L1–L5. Electron-donating effect descended the energy difference between HOMO and LUMO, leading to λem red shifted.43
 |
| | Fig. 7 (a) Emission spectra of Zn1–Zn5 in the solid state and (b) CIE chromaticity diagram (1931 CIE standard). (c) Emission spectra of Zn1–Zn5 in acetonitrile solution and (d) CIE chromaticity diagram. | |
In acetonitrile solution, the emission bands are observed at 393–413 nm, and 400–436 nm, respectively, for ligands L1–L5 (Fig. S9c and d†) and complexes Zn1–Zn5 (Fig. 7c and d). These bands exhibit deep blue luminescence emissions, and the Commission Internationale d'Eclairage (CIE) coordinates are summarized in Table 3. In acetonitrile solution, the maxima emission peaks are blue-shifted 65–87 nm compared with those in the solid state. These phenomena can be explained that the formation of the hydrogen bonds and π⋯π stacking interactions in the solid state, which can effectively decrease the HOMO–LUMO energy gap, and influence the ligand-centered π*–π transitions.44 Meanwhile, the full width at half-maximum (FWHM) of the emission band decreases from acetonitrile solution to the solid state (Table 3). All luminescence quantum yields of complexes Zn1–Zn5 in solution were measured by the optical dilute method of Demas and Crosby45 with a standard of quinine sulfate (Φr = 0.546) and are listed in Table 3. The luminescence quantum yield of Zn1–Zn5 in CH3CN is 0.095, 0.160, 0.193, 0.362 and 0.278, respectively, which is higher than that of the corresponding ligand. Particularly, the quantum yields of Zn4 (ΦF = 0.362) is 7.87-fold to L4 (ΦF = 0.046). This is because the ligands coordinated with zinc(II) ions increase the rigidity of the molecular edifice and reduce the loss of energy by radiationless thermal vibrations.46
The luminescence decay profiles of ligands L1–L5 and the corresponding zinc(II) complexes were measured at their optimal excitation wavelengths in the solid state and acetonitrile solution at 298 K (Fig. S10 and S11†). The detailed data are listed in Table S4.† A general trend is that the luminescence lifetimes for complexes Zn1–Zn5 either in the solid state or acetonitrile solution at 298 K are mostly longer than that of the corresponding ligands L1–L5. The most obvious observation is that the lifetime of Zn1 (τ = 9.15 μs) is 1.5-fold to the corresponding ligand L1 (τ = 6.10 μs) in acetonitrile solution. This is attributed to the more stable structure and interaction upon coordination.47 Meanwhile, the luminescence lifetimes of the ligand and corresponding complexes in the solid state (τ = 7.55–8.91 μs for L1–L5; τ = 8.37–10.25 μs for Zn1–Zn5) are longer than those in acetonitrile solution (τ = 6.10–8.62 μs for L1–L5; τ = 8.04–9.15 μs for Zn1–Zn5), which might be explained by the fact that there is a less polar nature in the solid-state environment.48
DFT calculations
The orbital distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy of Zn1–Zn5 were calculated by DFT/B3LYP/6-31G (Fig. 8). Their optimized molecular structures are obtained from X-ray diffraction analyses of Zn1–Zn5. Therefore, we obtain information about their electron density distributions, energy gaps, and energy levels of HOMO and LUMO based on the calculated optimized molecular structures.
 |
| | Fig. 8 Theoretically calculated spatial distributions and energies of the HOMO and LUMO levels of Zn1–Zn5. | |
For all selected complexes, the HOMO electron density is mainly placed on the phenyl ring, while the LUMO electron density is mainly located on the pyridyl ring as shown in Fig. 8. Indeed, an analysis of the orbital distributions reveals a lesser participation of the Zn atom in the emission process for all complexes. These facts further demonstrate the π–π* nature of the intraligand charge transfer transitions in each case. The HOMO–LUMO gap is found to be 4.06 eV for Zn1 (H) and decreases to 3.99 to 3.94 to 3.70 to 3.80 eV for Zn2 (2-Me) to Zn3 (2,4-Me2) to Zn4 (2,4,6-Me3) to Zn5 (2-OMe), respectively. These results are consistent with the fact of electron-donating effect descended the energy difference between HOMO and LUMO.
Conclusion
In summary, five Zn(II) complexes with different alkyl substituent were synthesized and the effect of alkyl substitution on the structural as well as spectroscopic properties were also examined. It was found that complexes Zn1–Zn5 exhibit 3D → 1D supramolecular frameworks in pace with an increase in the number of alkyl substitution. The emission maximum wavelengths of complexes Zn1–Zn5 can be tuned in a large range of 400–514 nm due to the substitution with different alkyl substituent. This influence of ligand modification on the properties of the complexes was also confirmed by the theoretical calculations (DFT). The luminescent properties of zinc Schiff base complex are easily tunable by variation of the alkyl substitution. These kinds of zinc Schiff base complexes with supramolecular framework shows great promise for the preparation of a wide variety of transition-metal complexes with predictable and tunable properties.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant 21371040 and 21571042), the National Key Basic Research Program of China (973 Program, No. 2013CB632900).
References
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- W. Y. Wong, L. Liu and J. X. Shi, Angew. Chem., Int. Ed., 2003, 42, 4064–4068 CrossRef CAS PubMed.
- W. Y. Wong, Coord. Chem. Rev., 2005, 249, 971–997 CrossRef CAS.
- L. L. Han, T. P. Hu, K. Mei, Z. M. Guo, C. Yin, Y. X. Wang, J. Zheng, X. P. Wang and D. Sun, Dalton Trans., 2015, 6052–6061 RSC.
- S. Yuan, Y. K. Deng and D. Sun, Chem.–Eur. J., 2014, 20, 10093–10098 CrossRef CAS PubMed.
- W. G. Lu, Z. W. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. W. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- L. Fu, M. Pan, X. J. Kuang, C. Yan, K. Li, S. C. Wei and C. Y. Su, J. Mater. Chem. A, 2013, 1, 8575–8580 CAS.
- X. L. Hu, C. Qin, X. L. Wang, K. Z. Shao and Z. M. Su, Chem. Commun., 2015, 51, 17521–17524 RSC.
- Y. Q. Jiao, H. Y. Zang, X. L. Wang, E. L. Zhou, B. Q. Song, C. G. Wang, K. Z. Shao and Z. M. Su, Chem. Commun., 2015, 51, 11313–11316 RSC.
- Q. Y. Yang, M. Pan, S. C. Wei, K. Li, B. B. Du and C. Y. Su, Inorg. Chem., 2015, 54, 5707–5716 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS.
- A. Ali, G. Hundal and R. Gupta, Cryst. Growth Des., 2012, 12, 1308–1319 CAS.
- D. Natale and J. C. Mareque-Rivas, Chem. Commun., 2008, 425–437 RSC.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC.
- J. S. Field, O. Q. Munro and B. P. Waldron, Dalton Trans., 2012, 5486–5496 RSC.
- J. M. Lin, Y. X. Qiu, W. B. Chen, M. Yang, A. J. Zhou, W. Dong and C. E. Tian, CrystEngComm, 2012, 14, 2779–2786 RSC.
- R. F. Semeniuc, T. J. Reamer and M. D. Smith, New J. Chem., 2010, 34, 439–452 RSC.
- G. Mahmoudi, V. Stilinović, M. S. Gargari, A. Bauzá, G. Zaragoza, W. Kaminsky, V. Lynch, D. Choquesillo-Lazarte, K. Sivakumar, A. A. Khandari and A. Frontera, CrystEngComm, 2015, 17, 3493–3502 RSC.
- W. K. Lo, W. K. Wong, W. Y. Wong and J. P. Guo, Eur. J. Inorg. Chem., 2005, 3950–3954 CrossRef CAS.
- Q. H. Meng, P. Zhou, F. Song, Y. B. Wang, G. L. Liu and H. Li, CrystEngComm, 2013, 15, 2786–2790 RSC.
- G. M. Sheldrick, SHELXTL NT Crystal Structure Analysis Package, Version 5.10, Analytical X-ray System, Bruker AXS, Madison, WI, 1999 Search PubMed.
- H. Huang, Y. Wang, B. Wang, S. Zhuang, B. Pan, X. Yang, L. Wang and C. Yang, J. Mater. Chem. C, 2013, 1, 5899–5908 RSC.
- C. Han, G. Xie, J. Li, Z. Zhang, H. Xu, Z. Deng, Y. Zhao, P. Yan and S. Liu, Chemistry, 2011, 17, 8947–8956 CrossRef CAS PubMed.
- Y. Yuan, J. X. Chen, F. Lu, Q. X. Tong, Q. D. Yang, H. W. Mo, T. W. Ng, F. L. Wong, Z. Q. Guo, J. Ye, Z. Chen, X. H. Zhang and C. S. Lee, Chem. Mater., 2013, 25, 4957–4965 CrossRef CAS.
- P. Baran, R. Boca, M. Breza, H. Elias, H. Fuess, V. Jorik, R. Klement and I. Svoboda, Polyhedron, 2002, 21, 1561–1571 CrossRef CAS.
- X. Gong, Y. Y. Ge, M. Fang, Z. G. Gu, S. R. Zheng, W. S. Li, S. J. Hu, S. B. Li and Y. P. Cai, CrystEngComm, 2011, 13, 6911–6915 RSC.
- M. Shakir, M. Azam, Y. Azim, S. Parveen and A. U. Khan, Polyhedron, 2007, 26, 5513–5518 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- Y. W. Dong, R. Q. Fan, P. Wang, L. G. Wei, X. M. Wang, S. Gao, H. J. Zhang, Y. L. Yang and Y. L Wang, Inorg. Chem., 2015, 54, 7742–7752 CrossRef CAS PubMed.
- M. Franks, A. Gadzhieva, L. Ghandhi, D. Murrell, A. J. Blake, E. S. Davies, W. Lewis, F. Moro, J. McMaster and M. Schröder, Inorg. Chem., 2013, 52, 660–670 CrossRef CAS PubMed.
- Y. W. Dong, R. Q. Fan, X. M. Wang, P. Wang, H. J. Zhang, L. G. Wei, W. Chen and Y. L. Yang, Cryst. Growth Des., 2016, 16, 3366–3378 CAS.
- F. Wu, H. Tong, Z. Li, W. Lei, L. Liu, W. Y. Wong, W. K. Wong and X. Zhu, Dalton Trans., 2014, 12463–12466 RSC.
- F. Wu, J. Li, H. Tong, Z. Li, C. Adachi, A. Langlois, P. D. Harvey, L. Liu, W. Y. Wong, W. K. Wong and X. Zhu, J. Mater. Chem. C, 2015, 3, 138–146 RSC.
- R. Y. Huang, H. Jiang, C. H. Zhu and H. Xu, RSC Adv., 2016, 6, 3341–3349 RSC.
- C. X. Ding, X. Rui, C. Wang and Y. S. Xie, CrystEngComm, 2014, 16, 1010–1019 RSC.
- X. Shi, G. S. Zhu, Q. R. Fang, G. Wu, G. Tian, R. W. Wang, D. L. Zhang, M. Xue and S. L. Qiu, Eur. J. Inorg. Chem., 2004, 185–191 CrossRef.
- C. Y. Xu, Q. Q. Guo, X. J. Wang, H. W. Hou and Y. T. Fan, Cryst. Growth Des., 2011, 11, 1869–1879 CAS.
- B. X. Dong and Q. Xu, Cryst. Growth Des., 2009, 9, 2776–2782 CAS.
- Y. H. Wu, L. Chen, J. Yu, S. L. Tong and Y. Yan, Dyes Pigm., 2013, 97, 423–428 CrossRef CAS.
- K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119–4130 CrossRef CAS PubMed.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- C. Maxim, F. Tuna, A. M. Madalan, N. Avarvari and M. Andruh, Cryst. Growth Des., 2012, 12, 1654–1665 CAS.
- H. Mandal, S. Chakrabartty and D. Ray, RSC Adv., 2014, 4, 65044–65055 RSC.
- K. D. Murugan and P. Natarajan, Eur. Polym. J., 2011, 47, 1664–1675 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 








