
Carbon–carbon vs. carbon–oxygen bond activation in 2- and 3-furonitriles with nickel†
Abstract
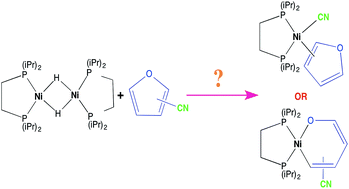
The reaction between [(dippe)NiH]2 (1) (dippe = 1,2-bis(diisopropylphosphino)-ethane) and 2- and 3-furonitrile (2-FN and 3-FN) was carried out at room temperature. Both furonitriles initially react with H2 loss and coordinating a [Ni(dippe)] fragment to the nitrile substituent in a η2-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N way to yield complexes [(dippe)Ni(η2-C,N-2-FN)] (2) and [(dippe)Ni(η2-C,N-3-FN)] (10), respectively. After monitoring this reaction for 6 days at room temperature in the presence of a slight excess of each nitrile, the C–CN bond activation products [(dippe)Ni(CN)(η1-C-2-FN)] (7) and [(dippe)Ni(CN)(η1-C-3-FN)] (11) were observed as the main products. These complexes were fully characterized by 1H, 31P{1H} and 13C{1H} NMR and by single crystal X-ray determinations. After isolation and characterization of (7) and (11), a thermal stability study was carried out, and it was found that the –C–CN bond activation is reversible at 100 °C for both compounds. Particularly, the thermolysis of complex (7) allowed the further production of a C–O ring opening at the furan to yield complex [(dippe)Ni(κ2-O,C–OCH
N way to yield complexes [(dippe)Ni(η2-C,N-2-FN)] (2) and [(dippe)Ni(η2-C,N-3-FN)] (10), respectively. After monitoring this reaction for 6 days at room temperature in the presence of a slight excess of each nitrile, the C–CN bond activation products [(dippe)Ni(CN)(η1-C-2-FN)] (7) and [(dippe)Ni(CN)(η1-C-3-FN)] (11) were observed as the main products. These complexes were fully characterized by 1H, 31P{1H} and 13C{1H} NMR and by single crystal X-ray determinations. After isolation and characterization of (7) and (11), a thermal stability study was carried out, and it was found that the –C–CN bond activation is reversible at 100 °C for both compounds. Particularly, the thermolysis of complex (7) allowed the further production of a C–O ring opening at the furan to yield complex [(dippe)Ni(κ2-O,C–OCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHC(CN))] (9), which was also isolated and characterized by multinuclear NMR.
CH–CHC(CN))] (9), which was also isolated and characterized by multinuclear NMR.
 Please wait while we load your content...
Please wait while we load your content...

