
N-, Fe-Doped carbon sphere/oriented carbon nanofiber nanocomposite with synergistically enhanced electrochemical activities†
Li Zhang,
Wei Wan,
Qiang Wang,
Wen-Chao Lu,
Bei-Hua Hou and
Ping Chen*
School of Chemistry and Chemical Engineering, Anhui University, Hefei, Anhui 230601, P. R. China. E-mail: chenping@ahu.edu.cn
First published on 22nd September 2016
Abstract
Catalysts for the oxygen reduction reaction (ORR) are crucial in metal–air batteries, fuel cells and other electrochemical devices. Developing heteroatom-doped carbon catalysts with high performance and stability in alkaline media for practical application of metal–air batteries or in acidic media for practical application of proton exchange membrane fuel cells is still difficult and challenging. Here, we report the N-, Fe-doped carbon sphere/oriented carbon nanofiber nanocomposite (NFe-CS/CNF). N-, Fe-doped carbon spheres (NFe-CS) were derived from p-phenylenediamine and oriented N-, Fe-doped carbon nanofibers (NFe-CNF) were derived from o-phenylenediamine. 3.89 at% of N and 0.38 at% of Fe were doped in a typical product, which has high surface area. The typical product exhibits a high ORR activity and stability in acidic media and reveals superior stability and tolerance to methanol than Pt/C. In alkaline media, the typical product shows superior activity to Pt/C. The performance of a zinc–air full cell under real battery operation conditions indicates that the typical product is a competitive alternative to commercial PtC. The unique structure of NFe-CS/CNF could create strong interactions between the nanospheres and oriented nanofibers, promote charger transfer at the interface and improve the conductivity. This work provides a new fundamental understanding and design strategy for excellent carbon-based ORR catalysts.
Introduction
The electrochemical oxygen reduction reaction (ORR) is a critical process for many devices which relate to energy storage and conversion, such as metal–air batteries, fuel cells and other electrochemical devices.1,2 Sluggish ORR kinetics at the cathode, scarcity and high cost of Pt-based catalysts hinder their widespread commercialization.3 Recently, many works have focused on exploring nonprecious ORR catalysts with good activity, especially nitrogen-doped carbon materials because of their excellent performance and low cost.4,5 Numerous and ‘cheap’ carbon-based ORR catalysts with activity comparable to commercial Pt/C have already been demonstrated in alkaline media (often 0.1 M KOH). However, most studies have reported just its ‘half cell’ results from rotating disk electrode (RDE) based on the forced-convection theory.6 In practical applications of metal–air batteries, concentration of oxygen in 6 M KOH is 10 times lower than that in 0.1 M KOH for a half-cell, thus the half-cell result affords very limited information on practical full-cell performance.7,8 Therefore, developing nonprecious metal ORR electrocatalysts with high-performance in practical application of metal–air batteries is still a challenging work.9–11 In addition, though great progress has made in developing these nonprecious metal electrocatalysts in alkaline media, the catalysts often exhibit weak ORR performance and instability in acidic media. Electrocatalysts for ORR in acidic media are crucial in proton-exchange membrane fuel cells and other electrochemical devices, which can be run with acidic electrolytes.12 Developing the carbon-based catalysts with high performance and stability in acidic media is still difficult and challenging.13,14Recently, the N-doped carbon materials such as carbon nanotubes, mesoporous carbon, graphene and carbon nanotube/graphene hybrid have been prepared and exhibited high ORR activities.15–17 In order to improve ORR activity of the nitrogen-doped carbon, how to control the nitrogen doping types and balance the doping content is very important. Improving and balancing the content of active pyridinic- and graphic-nitrogens in the doped carbon can bring high performance of ORR catalysts.18 Adding certain transition-metal elements to the N-doped carbon materials to form the co-doped carbon materials can remarkably promote the ORR activity in acidic or alkaline media. In the past several years, C–N–Fe and C–N–Co catalysts have been quickly developed because of the high-performance from coordination of the transition-metal cations with the nitrogen.19,20 In order to obtain the improved activity, it is very important to carefully choose the suitable precursors (containing the transition-metal and nitrogen elements).12,13,21–23
Some reports have shown that electrocatalysts with three-dimension nanostructure can bring the excellent electrocatalytical activities because of their high surface area, big pore volume and merits of effectively accelerating reactant and electron transport. The catalysts (e.g. heteroatom-doped carbon, C–N–Fe, C–N–Co, carbon/metal oxides/carbides/nitrides nanocomposites) with three-dimension nanostructure have been reported and shown high performance.24,25 Recently, we have focused on three-dimension nonprecious metal ORR electrocatalysts. We reported that nitrogen-doped graphene/carbon nanotube nanocomposite exhibits the synergistically enhanced electrochemical activities for ORR.17 We also developed the interconnected N-doped carbon framework with Co/Co3O4 nanoparticles with high ORR activity.26 The N-, Fe- and Co-tridoped carbon nanotube/nanoporous carbon nanocomposite27 and the nanocomposite based on graphene, CMK3, nitrogen, and trace cobalt28 were developed to promote ORR activity. Nitrogen-doped nanoporous carbon nanosheets derived from plant biomass were prepared and we found that NH3 etching can produce the 3D-carbon material with high BET surface area and a number of micropores.29
Here, we reported a unique three-dimension N-, Fe-doped carbon spheres/oriented carbon nanofibers nanocomposite (NFe-CS/CNF) catalyst. The N-, Fe-doped carbon spheres (NFe-CS) were derived from the p-phenylenediamine and the oriented N-, Fe-doped carbon nanofibers (NFe-CNF) were derived from the o-phenylenediamine. NFe-CS/CNF creates strong interaction and larger interfacial between the nanospheres and nanofibers, which could promote the charger transfer at the interface and improve the conductivity. The possible mechanism for the formation of the new structure and the enhanced electrochemical performance were discussed. This paper proposes a new insight design strategy for the research area of the three-dimension nonprecious metal catalysts with excellent performance.
Experimental methods
Materials
FeCl3, H2SO4 and KOH were bought from Aladdin Industrial Corporation (China). p-Phenylenediamine and o-phenylenediamine were bought from Sinopharm Chemical Reagent Co. Ltd (China). Pt/C (20%) was bought from Johnson Matthey. Nafion was purchased from DuPont Company. All solutions were prepared using Milli-Q water.Preparation of NFe-CS-400
Typically, 8.0 g of p-phenylenediamine and 65.0 mL of distilled water were put into a beaker. Then, 4.0 g of FeCl3 was added together with magnetic stirring until a homogeneous solution was obtained. The solution was transferred into a stainless steel autoclave (100 mL). After the hydrothermal process (140 °C for 4 h), the black product was produced. After washing with distilled water by filtration, the carbonaceous product from p-phenylenediamine (denoted with P1) was obtained by freeze drying for 6 h. Then, 400 mg of the P1 was annealed in N2 atmosphere (99.99%) at 950 °C for 2 h with the heating rate of 5 °C min−1 and the cooling rate of 5 °C min−1 and the N and Fe-doped carbon sphere derived from p-phenylenediamine (NFe-CS-400) was obtained.Preparation of NFe-CNF-400
NFe-CNF-400 was prepared according to the same steps as NFe-CS-400. Typically, 8.0 g of o-phenylenediamine and 65.0 mL of distilled water were put into a beaker. Then, 4.0 g of FeCl3 was added together with magnetic stirring until a homogeneous solution was obtained. The solution was transferred into a autoclave (100 mL). After the hydrothermal process, the black product was obtained. After washing with distilled water by filtration, the carbonaceous product from o-phenylenediamine (denoted with P2) was obtained by freeze drying for 6 h. Then, 400 mg of the P2 was annealed in N2 atmosphere (99.99%) at 950 °C for 2 h with the heating rate of 5 °C min−1 and the cooling rate of 5 °C min−1 and the N and Fe-doped nanofiber derived from o-phenylenediamine (NFe-CNF-400) was achieved.Preparation of NFe-CS/CNF-200-200
Typically, 200 mg of the carbonaceous product from p-phenylenediamine (P1) and 200 mg of the carbonaceous product from o-phenylenediamine (P2) were mixed by grind process. Then the mixture was annealed in N2 atmosphere at 950 °C for 2 h with the heating rate of 5 °C min−1 and the cooling rate of 5 °C min−1 and the NFe-CS/CNF-200-200 was obtained. By adjusting the amount of P1 and P2, we can easily prepare the products of NFe-CS/NFe-CNF-150-250, NFe-CS/NFe-CNF-180-220, NFe-CS/CNF-200-200, NFe-CS/NFe-CNF-220-180 and NFe-CS/NFe-CNF-250-150.Characterization
We used a field emission scanning electron microanalyzer (Hitachi SU8010) to obtain the scanning electron microscopy (SEM) images. The samples were dropped and dried onto silicon substrates. X-ray photoelectron spectroscopic (XPS) measurements were performed on an X-ray photoelectron spectrometer (ESCALab MKII). A Micrometrics ASAP2020 analyzer (USA) was used to obtain the BET surface areas (P/P0 = 0.05–0.35) and pore volume. A desorption isotherm was used to calculate the pore volume. Raman spectra were obtained from an inVia-Reflex spectrometer (Renishaw) with a 532 nm laser excitation. X-ray diffraction patterns (XRD) of the products were obtained from an XD-3 X-ray diffractometer with Cu Kα radiation (λ = 0.154 nm). Infrared spectra were recorded on a Nicolet FTIR spectrometer using KBr pellets.Electrochemical measurements
Electrochemical activities of the samples were measured by a rotating disk electrode (RDE) at CHI750E electrochemical workstation (Shanghai Chenhua, China) in a three-electrode cell at room temperature. For each sample (including 20% Pt/C catalyst), the catalyst (5 mg) was dispersed in 1 mL (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v isopropanol/Milli-Q water) mixed solvent with 40 μL Nafion solution (5 wt%). Then, the mixture was ultrasonicated for 30 min to obtain the catalyst ink.30,31 We adhered the ink (8 μL) on the glassy carbon disk (PINE, 5 mm diameter), which was used as the working electrode (catalyst loading of 0.200 mg cm−2). For the three-electrode cell, in acidic media, and Ag/AgCl was used as reference and the oxygen reduction tests were carried out in O2-saturated 0.5 M H2SO4 solution. In alkaline media, Hg/HgO was used as reference, and the oxygen reduction tests were carried out in O2-saturated 0.1 M KOH solution.
:3 v/v isopropanol/Milli-Q water) mixed solvent with 40 μL Nafion solution (5 wt%). Then, the mixture was ultrasonicated for 30 min to obtain the catalyst ink.30,31 We adhered the ink (8 μL) on the glassy carbon disk (PINE, 5 mm diameter), which was used as the working electrode (catalyst loading of 0.200 mg cm−2). For the three-electrode cell, in acidic media, and Ag/AgCl was used as reference and the oxygen reduction tests were carried out in O2-saturated 0.5 M H2SO4 solution. In alkaline media, Hg/HgO was used as reference, and the oxygen reduction tests were carried out in O2-saturated 0.1 M KOH solution.
Koutecky–Levich plots were analyzed at various electrode potentials. We can achieve the number of electrons transferred (n) (by calculating the slopes of the linear fit lines) according to the Koutecky–Levich equation:14,29,32
 | (1) |
| B = 0.62 × n × F × CO2 × DO22/3 × ν−1/6 | (2) |
485 C mol−1), CO2 means the oxygen concentration (solubility), DO2 means the oxygen diffusion coefficient, and ν means the kinematic viscosity. CO2, DO2 and ν for 0.5 M H2SO4 solution are 1.1 × 10−6 mol cm−3, 1.80 × 10−5 cm2 s−1 and 0.01 cm2 s−1, respectively. CO2, DO2 and ν for 0.1 KOH solution are 1.2 × 10−6 mol cm−3, 1.90 × 10−5 cm2 s−1 and 0.01 cm2 s−1, respectively.33,34
We used the standard three-electrode system to calibrate the Ag/AgCl and Hg/HgO electrodes. In the system, the Pt wires were used as the working and counter electrodes. Before the investigation, electrolytes were pre-purged and saturated with high purity H2.
Electrochemical impedance spectroscopy (EIS) measurements were performed in N2-saturated 0.5 M H2SO4 with the frequencies range from 10 kHz to 0.1 Hz using IM6e electrochemical workstation (Zahner-Elektrik, Germany) at room temperature.
Preparation of an air electrode
The air electrode was prepared by the as-prepared catalysts 10 wt%, poly(tetrafluoroethylene) (PTFE) binder 30 wt% and activated carbon (Sigma Aldrich) 60 wt%.35 Firstly, each material was sonicated in Milli-Q water for 1 h and each suspension was mixed ultrasonically for 1 h to form a homogeneous suspension. Excess water was removed by filtering the suspension and the slurry was dried. The air electrode was manufactured by a kneading and rolling process to make the fixed thickness of air cathode (900 μm). Finally, Cu-foam as a current collector was attached to the back side of the air electrode.Zinc–air full cell assembly and measurement
For the zinc–air full cell test, the zinc–air single cells were used in these experiments.35 Zinc plate was used as the anode electrode and air electrode was used as the cathode electrode. The experiment was performed on IM6e electrochemical workstation (Zahner-Elektrik, Germany) with current densities varied from 0 to 250 mA cm−2. The galvanostatic discharge curves of zinc–air cells were recorded at 10 and 100 mA cm−2, respectively.Results and discussion
Scheme 1 illustrated the preparation of the NFe-CS/CNF-200-200. (1) The carbonaceous product from p-phenylenediamine (denoted with P1) was obtained from the polymerization of p-phenylenediamine through a hydrothermal process; (2) the carbonaceous product from o-phenylenediamine (denoted with P2) was obtained from the polymerization of o-phenylenediamine through a hydrothermal process; (3) 200 mg of P1 and 200 mg of P2 were mixed by grind process, then the mixture was annealed in N2 atmosphere and the NFe-CS/CNF-200-200 was obtained. By adjusting the amount of P1 and P2, we can easily prepare the products with different carbon spheres and nanofibers. In addition, when 400 mg of P1 was annealed in N2 atmosphere and the product was NFe-CS-400. And when 400 mg of P2 was annealed in N2 atmosphere and the product was NFe-CNF-400.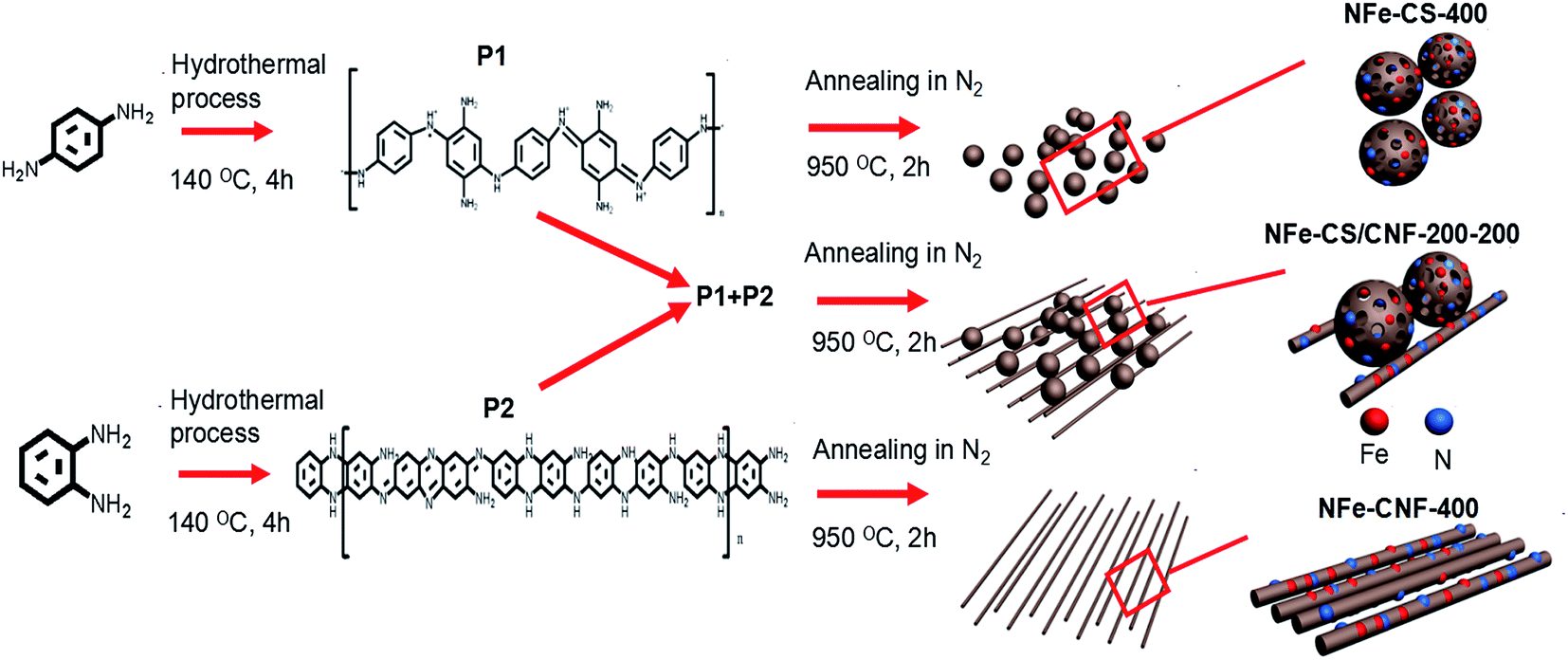 | ||
| Scheme 1 Schematic illustration of the preparation of the NFe-CS/CNF-200-200. | ||
Polymerization of p-phenylenediamine and o-phenylenediamine was confirmed by FTIR spectroscopy, respectively (ESI, Fig. S1†). Fig. S1a† shows the IR spectra of p-phenylenediamine and carbonaceous product from p-phenylenediamine (denoted with P1) in the 400–4000 cm−1 wavenumber range. In the spectrum of p-phenylenediamine, the peaks at 3376 cm−1 and 3304 cm−1 can be assigned to the N–H vibration. The peaks at 1630 cm−1 and 1515 cm−1 can be assigned to the existence of benzene ring. The intense peak at 826 cm−1 can indicate it is the substitutional aromatic compound. In the spectrum of the carbonaceous product from p-phenylenediamine (denoted with P1), the peaks at 1569 cm−1 appeared can be assigned to the quinone-type structure. These data confirm that the carbonaceous product (denoted with P1) is produced from polymerization of p-phenylenediamine. From the Fig. S1b,† the peaks at 3031 cm−1, 1600 cm−1, 1502 cm−1 and 750 cm−1 show the sample is o-phenylenediamine. The spectrum of the carbonaceous product from o-phenylenediamine (denoted with P2) shows the characteristic peaks at 1370 cm−1, 1226 cm−1 and 860 cm−1, indicating the polymerization of o-phenylenediamine.
Phenylenediamine has an amphiphilic structure of hydrophilic-NH2 and hydrophobic-C6H4, when phenylenediamine monomers are added to aqueous solution, droplets will be formed in the bulk solution. When FeCl3 is added to the solution, the polymerization only takes place at the water/droplet interface because of the hydrophilicity of FeCl3, which results in the formation of original nucleus.36 From the literature,37 the ladder-like structure conducted at temperatures between 800 and 1000 °C can easily obtain the fibers. The polymer of o-phenylenediamine has a ladder-like structure (P2), so the fibers can be formed after annealing at 950 °C. Because the polymer of p-phenylenediamine has not the ladder structure (P1), nanospheres are obtained.
Fig. 1a and b showed SEM image and magnified SEM image of NFe-CS/CNF-200-200. These clearly revealed that NFe-CS/CNF-200-200 contained the carbon nanoparticles with the diameter of 190–350 nm and carbon nanofibers with the diameter of 20–30 nm. The elemental mapping analysis (Fig. 1c–f) suggested the presence of C, N, O, and Fe components in NFe-CS/CNF-200-200. SEM image and elemental mapping analysis of NFe-CNF-400 were shown in Fig. S2 (see ESI†). The product contained the carbon nanofibers, and C, N, O, and Fe components were in NFe-CNF-400. SEM image and elemental mapping analysis of NFe-CS-400 were shown in Fig. S3 (see ESI†). The product contains the carbon nanoparticles with the diameter of 200–300 nm and C, N, O, and Fe components were in NFe-CS-400.
 | ||
| Fig. 1 (a–b) SEM image and magnified SEM image of NFe-CS/CNF-200-200; (c–f) carbon, nitrogen, oxygen and ferrum element mappings of NFe-CS/CNF-200-200. | ||
From XPS spectra of NFe-CS/CNF-200-200, the atomic percentage of C (91.73 at%), O (3.99 at%), N (3.89 at%) and Fe (0.38 at%) can be reached in the NFe-CS/CNF-200-200, respectively. From high-resolution N1s XPS spectra (Fig. 2a), the pyridinic-N (peak at 398.4 eV), pyrrolic-N (peak at 399.3 eV) and graphitic-N (peak at 401.1 eV) were doped in NFe-CS/CNF-200-200.30,31,38 The content of pyridinic- and graphic-nitrogens in the total nitrogen can approach to 96.35 at%. For the high performance of ORR catalysts, it is very important to improve and balance the content of active pyridinic- and graphic-nitrogens in the doped carbon.18 Fig. 2b and c indicate the pyridinic-N, pyrrolic-N and graphitic-N were doped in NFe-CS-400 and NFe-CNF-400. From Table S1,† N (3.54 at%) and Fe (0.28 at%) are doped in the NFe-CS-400, and N (4.32 at%) and Fe (0.72 at%) are doped in the NFe-CNF-400. From Fig. 2d and Table S1 (see ESI†), NFe-CS/CNF-200-200 exhibits high BET surface area of 625.00 m2 g−1, total pore volume of the 0.44 cm3 g−1 and micropore volume of the 0.28 cm3 g−1. And, from Fig. 2f and Table S1,† NFe-CNF-400 has BET surface area of 182.64 m2 g−1, total pore volume of the 0.22 cm3 g−1 and micropore volume of the 0.06 cm3 g−1. However, from Fig. 2e and Table S1,† NFe-CS-400 has BET surface area of 417.78 m2 g−1, total pore volume of 0.43 cm3 g−1 and micropore volume of the 0.17 cm3 g−1. Obviously, NFe-CS/CNF nanocomposite has higher surface area, pore volume than those of NFe-CS and NFe-CNF, respectively. X-ray diffraction of NFe-CS/CNF-200-200, NFe-CS-400 and NFe-CNF-400 (see ESI, Fig. S4a†) indicates the amorphous nature of carbon for the products.29,31 Raman spectra (Fig. S4b†) show that NFe-CS/CNF-200-200, NFe-CS-400 and NFe-CNF-400 exhibit the remarkable peaks at around 1350 and 1590 cm−1 corresponding to the well-defined D band and G band, respectively. In addition, a broader 2D peak appeared at around 2860 cm−1.29
 | ||
| Fig. 2 (a–c) The high-resolution N1s XPS spectra of NFe-CS/CNF-200-200, NFe-CS-400 and NFe-CNF-400; (d–f) nitrogen adsorption–desorption isotherms and pore distribution (insert) of the NFe-CS/CNF-200-200, NFe-CS-400 and NFe-CNF-400. | ||
ORR activities of samples in acidic media (0.5 M H2SO4) and alkaline media (0.1 M KOH) were investigated by RDE measurement, respectively. Commercial platinum (20 wt%) on carbon black (Pt/C) was also tested. ORR activities in 0.5 M H2SO4 were shown in Fig. 3a, from which the half-wave potential of NFe-CS-400, NFe-CNF-400, NFe-CS/CNF-200-200 and Pt/C was 525, 513, 782 and 850 mV (vs. RHE), respectively. Very remarkably, NFe-CS/CNF-200-200 electrode exhibits more positive half-wave potential and much larger reduction current than those of NFe-CS-400 and NFe-CNF-400. The half-wave potential of NFe-CS/CNF-200-200 is only 68 mV negative than that of Pt/C, and the current density of NFe-CS/CNF-200-200 is similar to that of Pt/C. We prepared the NFe-CS/CNF products with the different ratio of the carbonaceous product from p-phenylenediamine (denoted with P1) to the carbonaceous product from o-phenylenediamine (denoted with P2) (w/w) in preparation. Fig. 3b shows ORR polarization curves of NFe-CS/CNF-150-250, NFe-CS/CNF-180-220, NFe-CS/CNF-200-200, NFe-CS/CNF-220-180, NFe-CS/CNF-250-150. Noticeably, the ratio of product P1 to product P2 plays an important role in the activities. By controlling the appropriate ratio, ORR activities of the NFe-CS/CNF products can be remarkably promoted. NFe-CS/CNF-200-200 shows the best ORR activity than the other NFe-CS/CNF products. The above results demonstrate that the NFe-CS and NFe-CNF in NFe-CS/NFe-CNF have synergistically enhanced electrochemical activities for ORR. Fig. 3c shows the impedance data of NFe-CS-400, NFe-CNF-400, NFe-CS/NFe-CNF-150-250, NFe-CS/CNF-200-200 and NFe-CS/NFe-CNF-250-150. Nyquist plots of NFe-CS/CNF-200-200 exhibit very small charge transfer resistance in samples. Therefore, NFe-CS/CNF-200-200 is the most effective in shuttling charges from electrode to solution, and the catalytical activity for ORR can be remarkably promoted.17,39
 | ||
| Fig. 3 (a) ORR polarization curves in O2-saturated 0.5 M H2SO4 at room temperature (rotation speed 1600 rpm) for NFe-CS-400, NFe-CNF-400, NFe-CS/CNF-200-200 and Pt/C; (b) ORR polarization curves in O2-saturated 0.5 M H2SO4 for NFe-CS/CNF-150-250, NFe-CS/CNF-180-220, NFe-CS/CNF-200-200, NFe-CS/CNF-220-180, Fe-CS/CNF-250-150; (c) impedance data of the NFe-CS-400, NFe-CNF-400, NFe-CS/CNF-250-150, NFe-CS/CNF-200-200 and NFe-CS/CNF-150-250 in N2-saturated 0.5 M H2SO4; (d) ORR polarization curves for the ORR at the NFe-CS/CNF-200-200 electrode at the various rotation speeds; (e) current–time chronoamperometric response of the NFe-CS/CNF-200-200 and Pt/C electrodes at 0.75 V (versus RHE) at a rotation rate of 800 rpm; (f) ORR polarization curves at the NFe-CS/CNF-200-200 electrode with or without 1.5 M methanol. | ||
From polarization curves at NFe-CS/CNF-200-200 electrode with different rotation speeds (Fig. 3d) and K–L plots (i−1 vs. ω−1/2) at different electrode potentials (see ESI, Fig. S5a†), the number of electrons transferred was estimated to be 3.89–3.99, which implied a four-electron reduction process for ORR.40 Fig. 3e shows current–time (i–t) chronoamperometric response of NFe-CS/CNF-200-200 and Pt/C electrodes at 0.75 V (vs. RHE) in O2-saturated 0.5 M H2SO4 with a rotation rate of 800 rpm. After 19000 seconds, commercial Pt/C suffered from a 30.0% decrease in current density while NFe-CS/CNF-200-200 showed 14.2% loss of current density. NFe-CS/CNF-200-200 has a better durability than Pt/C in acidic media. From Fig. 3f and S5b† NFe-CS/CNF-200-200 has excellent tolerance to methanol. It is well-known that proton exchange membrane (PEM) fuel cells can be operated in acidic electrolytes. Therefore, NFe-CS/CNF-200-200 may have the potential application in practical application of PEM fuel cell.41
The ORR results in 0.1 M KOH were shown in Fig. 4a and b and S6 (see ESI†). From Fig. 4a, half-wave potential of NFe-CS-400, NFe-CNF-400, NFe-CS/CNF-200-200 and Pt/C was 739, 689, 863 and 845 mV (vs. RHE), respectively. Obviously, NFe-CS/CNF-200-200 electrode exhibits more positive half-wave potential and much larger reduction current than those of NFe-CS-400 and NFe-CNF-400. Remarkably, the onset potential and half-peak potential of NFe-CS/CNF-200-200 are positive than those of Pt/C, and the current density of NFe-CS/CNF-200-200 electrode is more than that of Pt/C. Fig. S6a† shows the Tafel plots of the NFe-CS/CNF-200-200 and Pt/C derived by the mass-transport correction of corresponding RDE data. From the Fig. S6a,† the NFe-CS/CNF-200-200 catalyst possesses a similar slope to the Pt/C, which indicates the facile ORR kinetics on the NFe-CS/CNF-200-200. From Fig. 4b, by controlling the ratio of product P1 to product P2, the half-peak potential and peak current of NFe-CS/CNF products can be remarkably elevated. NFe-CS/CNF-200-200 shows the best ORR activity in alkaline media. From RDE voltammograms at NFe-CS/CNF-200-200 electrode at various rotation speeds in 0.1 M KOH (Fig. S6b†) and K–L plots (Fig. S6c†), the number of electrons transferred was investigated to be 3.86–3.99, which revealed a four-electron reduction process for ORR in alkaline media. In addition, performance of directly mixed product of NFe-CS-400 and NFe-CNF-400 (200/200, w/w) for ORR was also measured (shown in Fig. S7†). Remarkably, NFe-CS/CNF-200-200 exhibits more positive half-wave potential and much larger reduction current than the directly mixed product.
 | ||
| Fig. 4 (a) ORR polarization curves in O2-saturated 0.1 M KOH (rotation speed 1600 rpm) for NFe-CS-400, NFe-CNF-400, NFe-CS/CNF-200-200 and Pt/C; (b) ORR polarization curves for NFe-CS/CNF-150-250, NFe-CS/CNF-180-220, NFe-CS/CNF-200-200, NFe-CS/CNF-220-180, NFe-CS/CNF-250-150; (c) photograph of the zinc–air single cell; (d) polarization curves (V–i) and corresponding power density plots of the batteries using NFe-CS/CNF-200-200 at different current density; (e and f) galvanostatic discharge curves of Zn–air battery with NFe-NPCS-900-4 and Pt/C as cathode catalysts at current density of 10 and 100 mA cm−2; (g and h) specific capacities of the Zn–air battery normalized to the mass of consumed Zn (at the current density of 10 and 100 mA cm−2) for NFe-NPCS-900-4 and Pt/C; (i) recharging the zinc–air single cell with NFe-CS/CNF-200-200 as cathode catalyst by changing Zn anode and electrolyte. The arrow stands for the recharging process. | ||
Fig. 3, 4a and b have shown the catalytical activities of NFe-CS/CNF-200-200 in alkaline and acidic media. Tables S2 and S3† provide the summary of reported ORR performance for nitrogen-doped carbon catalysts in alkaline and acidic media, respectively. Therefore, we can easily draw a conclusion that typical product in this work can show excellent catalytical activity for ORR in alkaline media and very high activity in acidic media.
Zinc–air (Zn–air) battery is a safe, resource-saving, environment-friendly energy device with a high specific energy.7,10,30,42 We characterized the performance of NFe-CS/CNF-200-200 in a Zn–air battery under real battery operation conditions. The zinc–air single cell was assembly (Fig. 4c), in which zinc plate was used as anode electrode and air electrode (including NFe-CS/CNF-200-200) was used as cathode electrode. For comparison, commercial Pt/C (20%) catalyst was also investigated as NFe-CS/CNF-200-200. The air electrode contains the as-prepared catalysts, poly(tetrafluoroethylene) (PTFE) binder and activated carbon. The electrode was manufactured by a kneading and rolling process to make the fixed thickness (900 μm). Cu-foam as a current collector was attached to the back side of the air electrode. Notably, in the Zn–air battery, electrolyte is 6.0 M KOH. Fig. 4d shows polarization curve (V–i) and corresponding power density plots of the battery using NFe-CS/CNF-200-200 at different current density. The maximum power density is 163.3 mW cm−2 when current density is 204 mA cm−2. While, from the polarization curve and corresponding power density plots of the battery using Pt/C (Fig. S8†), the maximum power density is 157.2 mW cm−2 when current density is 202 mA cm−2. From Fig. 4e and f, NFe-CS/CNF-200-200 shows the voltages of 1.29 and 1.04 V at the discharge current density of 10 mA cm−2 and 100 mA cm−2, respectively. For Pt/C, the voltages of 1.31 and 1.07 V was obtained at the discharge current density of 10 mA cm−2 and 100 mA cm−2, respectively. From the galvanostatic discharge curves, NFe-CS/CNF-200-200 has excellent durability. From Fig. 4g and h, the specific capacity of NFe-CS/CNF-200-200 was 717 mA h g−1 for 10 mA cm−2 and 656 mA h g−1 for 100 mA cm−2. For Pt/C, the specific capacity was 706 mA h g−1 for 10 mA cm−2 and 691 mA h g−1 for 100 mA cm−2. Performance of Zn–air battery using NFe-CS/CNF-200-200 was superior to most reported electrocatalysts (ESI, Table S4†). Especially, at a high current density (100 mA cm−2), the NFe-CS/CNF-200-200 revealed excellent performance of Zn–air battery with high voltage and specific capacity.10,35,43,44 NFe-CS/CNF-200-200 is a competitive alternative to commercial Pt/C in practical application of Zn–air battery. Fig. 4i shows the battery can be regenerated by changing Zn anode and electrolyte periodically.
From the above results and discussion, there are probably three reasons for the excellent ORR activity and the performance of Zn–air battery. Firstly, the high content of pyridinic N and graphitic N doped in catalyst can bring the high ORR activity.18,30,31 Co-doping of the N and Fe elements probably leads to the formation of the N–Fe bond, which plays a key role in ORR activity.45–47 Secondly, because of the orientation of the CNFs in NFe-CS/CNF nanocomposite (see from Fig. 1a), the CNFs may have higher electron transfer efficiency. NFe-CS/CNF contains the NFe-CS and NFe-CNF. The structure of NFe-CS/CNF could create strong interaction between the nanospheres and nanofibers, thus promoting the charger transfer at the interface and improving the conductivity. The impedance data of NFe-CS/CNF, NFe-CS and NFe-CNF can prove this.17 Thirdly, the NFe-CS/CNF-200-200 has the three-dimension nanostructure with the high BET surface area of (625.00 m2 g−1), total pore volume (0.44 cm3 g−1) and micropore volume (0.28 cm3 g−1), which brings many catalytic sites for ORR on the surface, and the active sites can be easily exposed to react.24,29 The three-dimension nanostructure also has the merits of effectively accelerating reactant and electron transport.17
Conclusion
A unique three-dimension NFe-CS/CNF catalyst was prepared. Typical product exhibits a high ORR activity and stability in acidic media. In alkaline media, typical product shows superior activity than commercial Pt/C and similar performance in the measurement of Zn–air full cell to Pt/C, which implied the typical product was a competitive alternative to Pt/C in practical application of zinc–air battery. Because of the orientation of the CNF in NFe-CS/CNF nanocomposite, the structure of NFe-CS/CNF could create strong interaction between the nanospheres and nanofibers, thus promote the charger transfer at the interface and improve the conductivity. The NFe-CS and NFe-CNF in the nanocomposite have synergistically enhanced electrochemical activities for ORR. The rational strategy would not only open up a new avenue boosting the electrocatalytic performance of carbon-based catalysts, but also give the inspiration for the design of other materials in the fields of supercapacitors, lithium ion batteries, sensors and so on. The preparation method was new, very efficient and scalable due to not using complex synthetic route.Acknowledgements
The authors are deeply grateful for the foundation: National Natural Science Foundation of China (21271005), Foundation of Anhui University (02303203-0054) and Foundation of Education Department of Anhui Province (gxyqZD2016010).References
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed
.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed
- R. B. Levy and M. Boudart, Science, 1973, 181, 547 CAS
- L. M. Dai, Y. H. Xue, L. T. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823 CrossRef CAS PubMed
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594 CrossRef CAS PubMed
- M. Zhou, H. L. Wang and S. J. Guo, Chem. Soc. Rev., 2016, 45, 1273 RSC
- Y. G. Li and H. J. Dai, Chem. Soc. Rev., 2014, 43, 5257 RSC
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746 RSC
- Z. Chen, A. P. Yu, D. Higgins, H. Li, H. J. Wang and Z. W. Chen, Nano Lett., 2012, 12, 1946 CrossRef CAS PubMed
- Y. G. Li, M. Gong, Y. Y. Liang, J. Feng, J. E. Kim, H. L. Wang, G. S. Hong, B. Zhang and H. J. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed
- H. W. Liang, W. Wei, Z. S. Wu, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002 CrossRef CAS PubMed
- L. Lin, Q. Zhu and A. W. Xu, J. Am. Chem. Soc., 2014, 136, 11027 CrossRef CAS PubMed
- Q. Wang, Z. Y. Zhou, Y. J. Lai, Y. You, J. G. Liu, X. L. Wu, E. Terefe, C. Chen, L. Song, M. Rauf, N. Tian and S. G. Sun, J. Am. Chem. Soc., 2014, 136, 10882 CrossRef CAS PubMed
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394 CrossRef CAS PubMed
- P. Chen, T. Y. Xiao, Y. H. Qian, S. S. Li and S. H. Yu, Adv. Mater., 2013, 25, 3192 CrossRef CAS PubMed
- D. H. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361 CrossRef CAS PubMed
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71 CrossRef CAS PubMed
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS PubMed
- Z. L. Li, G. L. Li, L. H. Jiang, J. L. Li, G. Q. Sun, C. G. Xia and F. W. Li, Angew. Chem., Int. Ed., 2015, 54, 1494 CrossRef CAS PubMed
- E. Proietti, F. Jaouen, M. Lefevre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 109 Search PubMed
- L. Wenmu, Y. Aiping, D. C. Higgins, B. G. Llanos and C. Zhongwei, J. Am. Chem. Soc., 2010, 132, 17056 CrossRef PubMed
- C. Z. Zhu, H. Li, S. F. Fu, D. Du and Y. H. Lin, Chem. Soc. Rev., 2016, 45, 517 RSC
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed
- Z. Y. Wu, P. Chen, Q. S. Wu, L. F. Yang, Z. Pan and Q. Wang, Nano Energy, 2014, 8, 118 CrossRef CAS
- G. Wang, W. H. Wang, L. K. Wang, W. T. Yao, P. F. Yao, W. K. Zhu, P. Chen and Q. S. Wu, J. Mater. Chem. A, 2015, 3, 17866 CAS
- X. J. Huang, Y. G. Tang, L. F. Yang, P. Chen, Q. S. Wu and Z. Pan, J. Mater. Chem. A, 2015, 3, 2978 CAS
- P. Chen, L. K. Wang, G. Wang, M. R. Gao, J. Ge, W. J. Yuan, Y. H. Shen, A. J. Xie and S. H. Yu, Energy Environ. Sci., 2014, 7, 4095 CAS
- W. Wan, Q. Wang, L. Zhang, H. W. Liang, P. Chen and S. H. Yu, J. Mater. Chem. A, 2016, 4, 8602 CAS
- W. T. Yao, L. Yu, P. F. Yao, K. Wei, S. L. Han, P. Chen and J. S. Xie, ACS Sustainable Chem. Eng., 2016, 4, 3235 CrossRef CAS
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed
- R. E. Davis, G. L. Horvath and C. W. Tobias, Electrochim. Acta, 1967, 12, 287 CrossRef CAS
- M. K. Tham, R. D. Walker and K. E. Gubbins, J. Phys. Chem., 1970, 74, 1747 CrossRef CAS
- J. S. Lee, G. S. Park, H. I. Lee, S. T. Kim, R. G. Cao, M. L. Liu and J. Cho, Nano Lett., 2011, 11, 5362 CrossRef CAS PubMed
- J. Han, G. Song and R. Guo, Eur. Polym. J., 2007, 43, 4229 CrossRef CAS
- L. T. Memetea, N. C. Billingham and E. T. H. Then, Polym. Degrad. Stab., 1995, 47, 189 CrossRef CAS
- W. Niu, L. Li, X. Liu, N. Wang, J. Liu, W. Zhou, Z. Tang and S. Chen, J. Am. Chem. Soc., 2015, 137, 5555 CrossRef CAS PubMed
- Y. Y. Liang, H. L. Wang, P. Diao, W. Chang, G. S. Hong, Y. G. Li, M. Gong, L. M. Xie, J. G. Zhou, J. Wang, T. Z. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 15849 CrossRef CAS PubMed
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS PubMed
- C. Medard, M. Lefevre, J. P. Dodelet, F. Jaouen and G. Lindbergh, Electrochim. Acta, 2006, 51, 3202 CrossRef CAS
- J. Fu, D. U. Lee, F. M. Hassan, L. Yang, Z. Y. Bai, M. G. Park and Z. W. Chen, Adv. Mater., 2015, 27, 5617 CrossRef CAS PubMed
- H. W. Liang, X. Zhuang, S. Brüller, X. L. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed
- H.-W. Liang, Z.-Y. Wu, L.-F. Chen, C. Li and S.-H. Yu, Nano Energy, 2015, 11, 366 CrossRef CAS
- J. Liu, X. J. Sun, P. Song, Y. W. Zhang, W. Xing and W. L. Xu, Adv. Mater., 2013, 25, 6879 CrossRef CAS PubMed
- X. Zhong, L. Liu, X. D. Wang, H. Y. Yu, G. L. Zhuang, D. H. Mei, X. N. Li and J. G. Wang, J. Mater. Chem. A, 2014, 2, 6703 CAS
- X. Zhong, H. Yu, G. Zhuang, Q. Li, X. Wang, Y. Zhu, L. Liu, X. Li, M. Dong and J. Wang, J. Mater. Chem. A, 2013, 2, 897 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20075c |
| This journal is © The Royal Society of Chemistry 2016 |