
Diverse acetals from stoichiometric amounts of aldehydes and alcohols under very mild conditions: a new twist to PPh3–CCl4 reagent combination†
Abstract
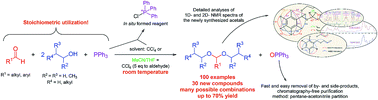
The most frequently utilized method for the preparation of acetals is the reaction of alcohols and aldehydes or ketones, but it suffers from a number of shortcomings: bad thermodynamics, the presence of an acid catalyst and occasionally a complicated reaction workup. Frequently, PPh3 in combination with CCl4 has been successfully used in the Appel reaction and for the preparation of esters and amides as a chlorinating and/or dehydrating agent. In this work, the possible use of PPh3–CCl4 reagent in the synthesis of acetals from aldehydes and alcohols was studied. By varying the ratio of reactants and reaction conditions (temperature, reaction time, solvent), the reaction yield was optimized, while side products were reduced to a minimum. The best results were attained when a near stoichiometric ratio (1 : 2.2 : 1.1 = aldehyde/alcohol/PPh3) of the reactants was used and the reaction was conducted at room temperature in CCl4. This reagent ratio also resulted in reaction mixtures that besides OPPh3, residual reactants and the acetal, contained no more than 10% of side products, with respect to the acetal. Chromatography-free acetal isolation/purification was readily accomplished using a pentane–acetonitrile partition. In this way we obtained 100 acetals out of which 30 acetals represented new compounds which were fully spectrally (1D-, 2D-NMR, MS, IR, UV) characterized. The reaction mechanism behind this newly developed synthetic procedure was elucidated and discussed.
 Please wait while we load your content...
Please wait while we load your content...

