
HCl removal performance of Mg-stabilized carbide slag from carbonation/calcination cycles for CO2 capture
Changyun Chia,
Yingjie Li*a,
Rongyue Sunb,
Xiaotong Maa,
Lunbo Duanb and
Zeyan Wangc
aSchool of Energy and Power Engineering, Shandong University, Jinan, 250061, China. E-mail: liyj@sdu.edu.cn
bKey Laboratory of Energy Thermal Conversion and Control, Ministry of Education, School of Energy and Environment, Southeast University, Nanjing, 210096, China
cState Key Laboratory of Crystal Materials, Shandong University, Jinan, 250100, China
First published on 18th October 2016
Abstract
Mg-stabilized carbide slag (MSCS) was fabricated with carbide slag, magnesium nitrate hydrate and a by-product of biodiesel from transesterification by combustion, and was used as a CO2 sorbent in calcium looping cycles. The cycled MSCS containing a ratio of CaO to MgO of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 from the calcium looping cycles (i.e. carbonation/calcination cycles) for CO2 capture was subsequently used as an HCl sorbent. The HCl capture performance of the cycled MSCS which had experienced repetitive CO2 capture cycles using calcium looping was investigated in a triple fixed-bed reactor. The reaction products of the cycled MSCS after HCl absorption are CaClOH, CaO, and MgO. MgO is an inert support. The cycled MSCS reaches the highest HCl capture capacity at 750 °C. The number of CO2 capture cycles increases the effect of chlorination temperature on the HCl capture capacity of the cycled MSCS. The HCl capture capacity of the cycled MSCS drops slightly with the number of CO2 capture cycles. The HCl capture capacity of the MSCS which has undergone 10 CO2 capture cycles can retain 0.21 g HCl/g sorbent, which is about 1.7 times as high as that of the carbide slag. The presence of CO2 leads to a reduction in the HCl capture capacity of the cycled MSCS. MSCS can maintain a more stable microstructure due to the presence of MgO during the repetitive CO2 capture cycles. The cycled MSCS from the CO2 capture cycles exhibits a more porous structure than the cycled carbide slag, especially in the pore size range of 2–10 nm in diameter, which benefits HCl capture. Therefore, the cycled MSCS from CO2 capture cycles using calcium looping appears promising to remove HCl.
:20 from the calcium looping cycles (i.e. carbonation/calcination cycles) for CO2 capture was subsequently used as an HCl sorbent. The HCl capture performance of the cycled MSCS which had experienced repetitive CO2 capture cycles using calcium looping was investigated in a triple fixed-bed reactor. The reaction products of the cycled MSCS after HCl absorption are CaClOH, CaO, and MgO. MgO is an inert support. The cycled MSCS reaches the highest HCl capture capacity at 750 °C. The number of CO2 capture cycles increases the effect of chlorination temperature on the HCl capture capacity of the cycled MSCS. The HCl capture capacity of the cycled MSCS drops slightly with the number of CO2 capture cycles. The HCl capture capacity of the MSCS which has undergone 10 CO2 capture cycles can retain 0.21 g HCl/g sorbent, which is about 1.7 times as high as that of the carbide slag. The presence of CO2 leads to a reduction in the HCl capture capacity of the cycled MSCS. MSCS can maintain a more stable microstructure due to the presence of MgO during the repetitive CO2 capture cycles. The cycled MSCS from the CO2 capture cycles exhibits a more porous structure than the cycled carbide slag, especially in the pore size range of 2–10 nm in diameter, which benefits HCl capture. Therefore, the cycled MSCS from CO2 capture cycles using calcium looping appears promising to remove HCl.
1 Introduction
Mankind faces an increasing energy crisis and environmental pollution. China relies on fossil resources such as coal for the bulk of it’s energy, and emissions like SO2, NOx and CO2 are pumped into the atmosphere. Biomass and refuse derived fuel (RDF) are good substitute fuels, but their flue gas often contains a significant quantity of HCl. HCl concentration can reach 1900 ppm during combustion and gasification processes of biomass,1 and the HCl concentration in flue gas from RDF-fired boilers can reach levels even higher than 2500 ppm.2,3 HCl has a negative impact on the corrosion of the boiler heating surface and results in environmental pollution by polychlorinated dibenzo-dioxins (PCDDs) and furans (PCDFs).4–6 Thus, in order to take advantage of fuels containing chlorine such as biomass and RDF as substitutes for coal, it is essential to remove HCl efficiently during the application process of such fuels.Methods for HCl removal are commonly classified as “wet” and “dry”.7 In the dry method, calcium-based sorbents such as limestone and dolomite have often been used as HCl sorbents. Although the optimum reaction temperature for a chlorination reaction between CaO derived from various calcium-based sorbents and HCl may not yet be in full agreement, lots of references have reported that CaO has a high HCl capture capacity in a temperature range of 600–800 °C.8 Limestone can be used to capture HCl by direct chlorination (as is shown in eqn (1)) or indirect chlorination (chlorination after the decomposition of limestone as is shown in eqn (2) and (3)) at various temperatures.5 The indirect chlorination of limestone shows a higher HCl capture capacity than direct chlorination. CaO from different precursors other than limestone such as calcium hydroxide, dolomite, hydrogarnet, calcium magnesium acetate and calcium propionate exhibited a higher HCl capture capacity than that of limestone.4,5,9
| CaCO3 + 2HCl → CaCl2 + H2O + CO2 | (1) |
| CaCO3 → CaO + CO2 | (2) |
| CaO + 2HCl → CaCl2 + H2O | (3) |
Calcium-based sorbents are not only used to remove HCl, but are also used to capture CO2. CO2 capture and separation are regarded as potential methods to continue using fossil fuel-fired or biomass-fired power plants for electricity production, as ways to capture the main CO2 emission sources.10,11 Calcium looping using carbonation/calcination cycles of CaO is considered to be one of the most promising CO2 capture technologies for coal-fired power plants and hydrogen production.12 Calcium looping involves a carbonation reaction of CaO in a carbonator (650–700 °C) and a subsequent calcination reaction of the formed CaCO3 to regenerate CaO in a calciner (>850 °C), as is shown in eqn (4). Fuel is burnt during the oxy-combustion process in the calciner to produce the necessary heat for the regeneration of CaO. The almost pure CO2 stream from the calciner can be sequestrated or reused. Calcium looping can be applied in both pre-combustion CO2 capture and post-combustion CO2 capture,12–14 which has attracted a great deal of attention recently, owing to a number of advantages: the relatively small efficiency penalty, the potential use in large-scale circulating fluidized beds; it is an excellent opportunity for integration with cement manufacture.15
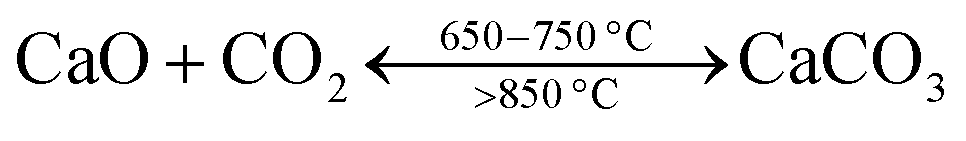 | (4) |
However, the CO2 capture capacity of the natural calcium-based sorbent decreases sharply with the number of cycles due to the sintering of CaO at high temperatures.16,17 Improving the CO2 capture capacity and cyclic stability of the calcium-based sorbent has become a research focus.18–23 The criterion for the design of the synthetic sorbent for CO2 capture is to increase the active surface area, stability of the pore structure, and mechanical stability of the sorbent, which enhances the CO2 capture capacity of the sorbent during calcium looping.24 Dispersing CaO into an inert carrier to weaken the sintering and aggregation of CaO particles has been frequently studied. The presented works have proved the effectiveness of adding various inert carriers such as Al2O3,25–27 ZrO2,28 SiO2,13,29 Y2O3,30 MnO2,31 CaTiO3,32 Nd2O3 (ref. 33) and calcium aluminate cement.34–39
Dolomite mainly composed of CaO and MgO shows a higher CO2 capture capacity than limestone during calcium looping cycles, because MgO as the inert carrier can hinder CaO sintering at high temperatures and maintain a stable pore structure during the cycles.40,41 MgO is a good support at high temperatures.42 MgO does not react with CO2 at calcium looping conditions. Some researchers have proposed MgO as an inert carrier to fabricate a synthetic CaO/MgO sorbent during calcium looping cycles. The CO2 capture performance of synthetic CO2 sorbents strongly depends on the synthesis routes and precursors19 and the cost should be considered for industrial applications. Broda et al.43 pointed out that MgO in the synthetic sorbent prevented the sintering of CaO particles and they used calcium acetate hydrate, alcohols, alkanes or toluene as raw materials through a “one-pot” re-crystallization method. Luo et al.44 adopted a wet mixing method to prepare CaO/MgO sorbents with calcium carbonate, calcium nitrate tetrahydrate, citric acid monohydrate, magnesium carbonate and magnesium nitrate hexahydrate. Liu et al.45,46 synthesized CaO/MgO sorbents through a wet mixing method and spray-drying method with calcium D-gluconate monohydrate and magnesium salts of D-gluconic acid, and the sorbents could achieve a CO2 capture capacity of 0.56 g CO2/g sorbent after 24 cycles and 0.46 g CO2/g sorbent after 44 cycles, respectively. Our group fabricated a novel synthetic CaO/MgO sorbent, i.e. Mg-stabilized carbide slag (MSCS), with carbide slag, magnesium nitrate hydrate and a by-product of biodiesel obtained from the transesterification process for biodiesel production.47 MSCS showed a high CO2 capture capacity, which could retain a CO2 uptake of 0.42 g/g sorbent after 20 cycles.
Carbide slag as an industrial waste is a by-product from the hydrolysis process of calcium carbide (CaC2) for the industrial production of ethylene as a main material of polyvinyl chloride.48 Xie et al.49 proposed a new HCl removal route where HCl can be removed by the cycled carbide slag from CO2 capture cycles using calcium looping. They used carbide slag and aluminum nitrate to fabricate CaO/Ca3Al2O6 sorbent and found that the cycled synthetic sorbent experienced 20 and 50 carbonation/calcination cycles for CO2 capture that were 2.3 and 2.6 times greater than those of the cycled carbide slag after the number of same cycles.50 HCl removal by sorbents from calcium looping that can realize the sequential capture of CO2 and HCl in the flue gas of fuels containing chlorine from fired power plants is promising.
Although our previous work47 has proved that the novel synthetic CO2 sorbent, i.e. MSCS prepared from carbide slag, magnesium nitrate hydrate and a by-product of biodiesel, possesses a high CO2 capture capacity and good stability during calcium looping cycles, the HCl capture performance of the cycled MSCS from CO2 capture cycles has not been reported and is necessary to be investigated.
In this work, the HCl capture performance of the cycled MSCS from CO2 capture cycles using calcium looping was studied. The effects of the chlorination temperature, the number of CO2 capture cycles, the HCl concentration and the presence of CO2 in the chlorination atmosphere on HCl removal by the cycled MSCS from calcium looping cycles were discussed. In addition, the HCl capture performance of the cycled MSCS was compared to that of the cycled carbide slag from CO2 capture cycles.
2 Experimental
2.1 Sample preparation
Carbide slag was sampled from a chlor-alkali plant in Shandong Province, China. The by-product of biodiesel (>90 wt% glycerol content) was obtained from the transesterification process of peanut oil reacted with methanol.51 The chemical components of the carbide slag analyzed by X-ray fluorescence (XRF) are shown in Table 1. The carbide slag with a particle size of less than 0.125 mm after being sieved was chosen for the following experiments.| CaO | MgO | SiO2 | Fe2O3 | Al2O3 | SrO | TiO2 | Others | LOI |
|---|---|---|---|---|---|---|---|---|
| 69.52 | 0.02 | 2.34 | 0.17 | 1.52 | 0.03 | 0.03 | 0.57 | 25.80 |
The preparation process for MSCS is presented as follows: 80 mL of the by-product of biodiesel and 50 mL of deionized water were added into a beaker and continuously stirred for 10 min. Then 5 g of carbide slag and 5.617 g of magnesium nitrate hydrate (analytical grade Mg(NO3)2·6H2O, > 99 wt%) were added to the by-product of biodiesel and stirred at 80 °C for 1 h to make them completely dissolve. The mass ratio of CaO derived from the carbide slag to MgO derived from magnesium nitrate hydrate was 80:20. Lastly, the solution was put into a muffle furnace for combustion under air at 850 °C for 1 h.
The synthetic sorbent, i.e. MSCS, was ground and sieved to a size of < 0.125 mm.
2.2 Triple fixed-bed reactor (TFBR)
The triple fixed-bed reactor (TFBR) mainly includes a calciner, a carbonator and a chlorinator, which is operated under atmospheric pressure, as is shown in Fig. 1. A gas flow rate of 2 L min−1 was controlled by the reducing valves and mass flow meters. The carbonator was operated at 700 °C for 20 min in 20% CO2/80% N2, and the calciner was controlled at 850 °C for 10 min in pure N2 (99.999%). The chlorinator was operated at 700–800 °C for 90 min in a gas mixture consisting of HCl (0.10–0.20%) and N2 (balance). 10% CO2 was added into the chlorination atmosphere in order to investigate the effect of the presence of CO2 on the HCl capture by the cycled MSCS. All of the gases were mixed in a mixer and the gas mixture was then sent to the TFBR.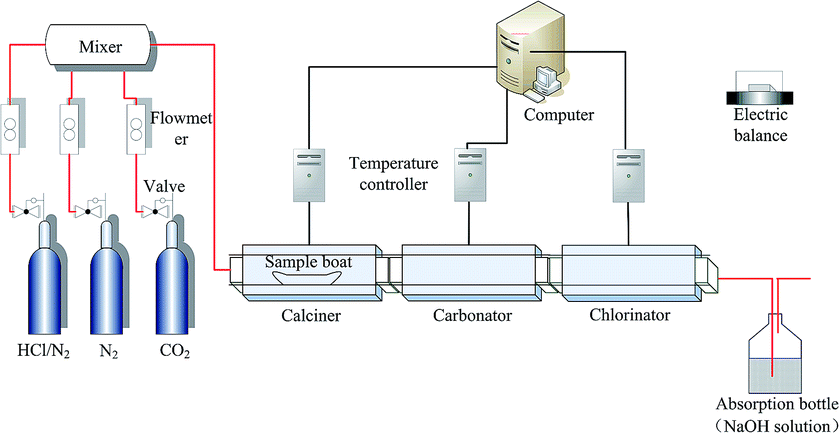 | ||
| Fig. 1 Schematic of the TFBR. | ||
500 mg of sample was evenly put into a boat where the thickness of the sample was less than 2 mm, and was firstly placed in the carbonator. After finishing carbonation, the sample boat was sent to the calciner for complete calcination. Then the 1st CO2 capture cycle, i.e. carbonation/calcination cycle, was finished. The calcined sample from the calciner was put into the carbonator again for the next CO2 capture cycle. The sample after 0 cycles denotes the initial sorbent which has not experienced carbonation/calcination cycles. The sorbent samples which had experienced 0, 1, 5, 10 and 20 CO2 capture cycles in the TFBR were sent into the chlorinator for HCl absorption. In the experimental process, the mass change of the sample was measured by an electronic balance (Mettler Toledo-XS105DU) with a resolution of 0.1 mg.
The HCl capture capacity can be calculated according to the mass change of the sample. The HCl capture capacity of the sample is defined as the HCl absorption amount per unit mass of the sample, which is presented as follows:
 | (5) |
It should be noted that each group of experiments has been repeated three times, and the accidental errors were all less than 2.8%. In order to show the results clearly, four figures were chosen to draw error bars.
2.3 Phase and microstructure analysis
The phase components of the cycled samples after 90 min of chlorination were analyzed by a D/Max-III A X-ray diffractometer (XRD). The surface morphologies of the cycled samples after different conditions were observed by a SUPRATM 55 field emission scan electron microscope (SEM). An Oxford INCA sight X energy dispersive spectrometer (EDS) was used to detect elements on the surfaces of the samples. The pore structure parameters of the samples were measured by a Micromeritics ASAP 2020-M nitrogen absorption analyzer. BET specific surface areas and pore size distributions of the samples were calculated according to the Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) model, respectively.3 Results and discussion
3.1 XRD analysis of chlorinated MSCS experiencing CO2 capture cycles
During the preparation process of MSCS, the mass ratio of CaO to MgO was chosen as 80:20, because our previous work proved that the obtained MSCS possessed the highest CO2 capture capacity during multiple cycles.47 Fig. 2 shows the XRD spectra of the chlorinated MSCS and the chlorinated carbide slag experiencing various CO2 capture cycles. The main products of the cycled carbide slag after 90 min of chlorination are CaO and CaClOH, as is shown in Fig. 2(a), which agrees with the results reported by Xie et al.49 The main components of MSCS are CaO and MgO,47 and the mass ratio of CaO to MgO is 80:20. The reaction products of MSCS after chlorination are CaO, MgO and CaClOH, as is presented in Fig. 2(b). This means that MgO as a support is inert and can not absorb HCl. CaCl2 is not generated during the 90 min chlorination of MSCS. Chin et al.9 and Partanen et al.52 both thought that CaClOH should be the first product generated when HCl was captured by CaO, but CaClOH ultimately converted to CaCl2 after a long reaction time (e.g. 800 min). However, HCl capture by the cycled MSCS from CO2 capture cycles may be applied in the fluidized bed reactors and the residence time of the sorbent in the reactor is short. Thus, the possible chlorination product of the cycled MCSC should be CaClOH. The chlorination reaction of MSCS after 0 and 10 cycles can be described as follow.| CaO·MgO + HCl → CaClOH·MgO | (6) |
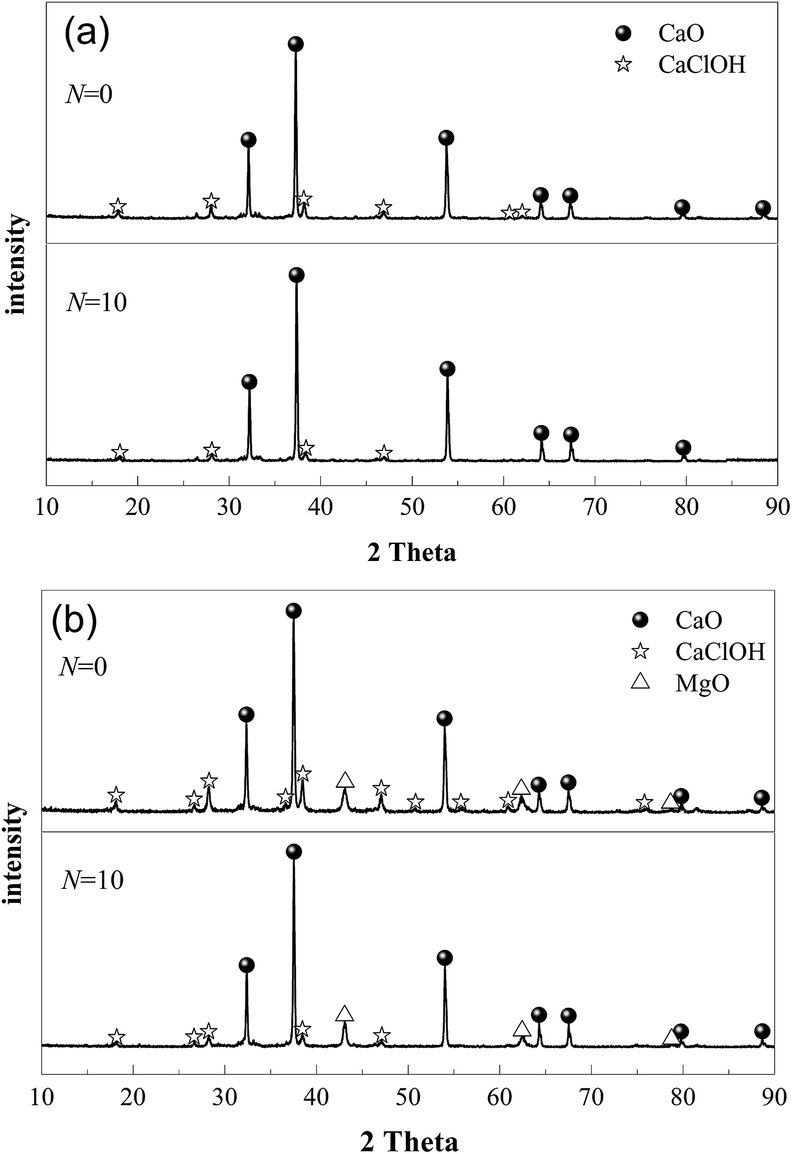 | ||
| Fig. 2 XRD spectra of chlorinated MSCS (a) and chlorinated carbide slag (b) from various CO2 capture cycles (chlorination at 750 °C in 0.20% HCl/N2 for balance, chlorination time: 90 min). | ||
3.2 Effect of the number of CO2 capture cycles on HCl capture by cycled MSCS
The effect of the number of CO2 capture cycles on the HCl capture performance of the cycled MSCS is depicted in Fig. 3. The HCl capture capacity and chlorination rate of the cycled MSCS are both obviously higher than those of the cycled carbide slag from the same CO2 capture cycles. It is found that the HCl capture capacity of the cycled MSCS linearly increases with chlorination time. As the number of CO2 capture cycles increases from 0 to 1, the HCl capture capacities of MSCS after 30 min and 90 min drop by about 30% and 11%, respectively. However, with increasing the cycle number from 1 to 20, the HCl capture capacities of the cycled MSCS only experience little change. Therefore, MSCS which has not experienced CO2 capture cycles achieves a higher HCl capture capacity, compared to the cycled MSCS during 20 cycles. The number of CO2 capture cycles exhibits little effect on YHCl,N of the cycled carbide slag during 20 cycles. After 30 min of chlorination, YHCl,0 and YHCl,10 of MSCS are about 2.5 and 2.2 times greater than those of the carbide slag, respectively. | ||
| Fig. 3 Effect of the number of CO2 capture cycles on YHCl,N of cycled MSCS from various CO2 capture cycles (chlorination at 750 °C in 0.20% HCl/N2 for balance). | ||
Fig. 4 presents a comparison between MSCS and the carbide slag, which have both experienced N CO2 capture cycles, of YHCl,N for 90 min of chlorination. Both the cycled MSCS and the cycled carbide slag exhibit relatively stable HCl capture capacity with an increasing number of CO2 capture cycles. The cycled MSCS achieves a much higher YHCl,N than the cycled carbide slag. YHCl,5 and YHCl,20 of MSCS are about 1.7 and 1.8 times higher than those of the carbide slag, respectively. This may be attributed to the support of MgO in MSCS. MSCS not only exhibits a higher cyclic CO2 capture capacity than the carbide slag,47 but shows a higher HCl capture capacity after CO2 capture cycles. Therefore, MSCS is a promising synthetic sorbent for sequential CO2/HCl capture.
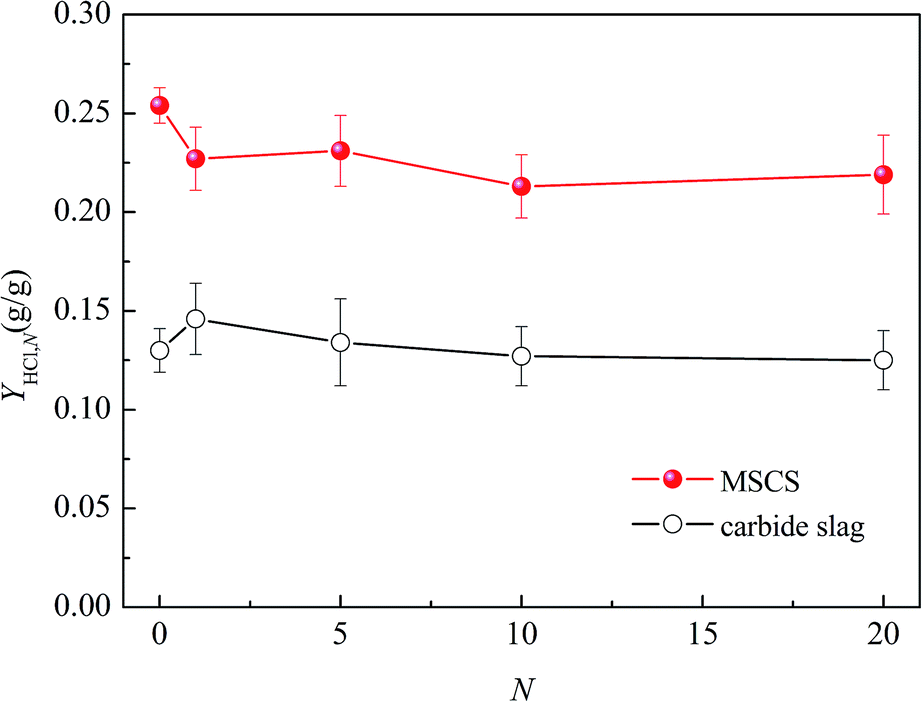 | ||
| Fig. 4 Comparison between MSCS and carbide slag from various CO2 capture cycles in YHCl,N for 90 min chlorination (chlorination at 750 °C in 0.20% HCl/N2 for balance). | ||
3.3 Effect of chlorination temperature on HCl capture by cycled MSCS
The effect of chlorination temperature in the range of 700–800 °C on the HCl capture capacity of the cycled MSCS under 90 min of chlorination is shown in Fig. 5. The HCl capture capacity of the cycled MSCS increases with increasing temperature from 700 to 750 °C, while it decays when further increasing the temperature to 800 °C. Therefore, the optimum chlorination temperature of the cycled MSCS from CO2 capture cycles is 750 °C. When the reaction temperature is low, a high temperature is beneficial for HCl absorption by CaO according to the chemical equilibrium between the gas and solid. However, when the reaction temperature is above 750 °C, the sintering of the sorbent is intensified which leads to the blockage and collapse of CaO in MSCS. That is not beneficial for HCl absorption by MSCS. Daoudi and Walters53 also found adverse effects of high-temperature sintering on HCl absorption by CaO. On the other hand, Weinell et al.54 found that at temperatures exceeding 750 °C, a liquid phase of chlorination product saturated with CaO was formed during the absorption of HCl by CaO, which is not beneficial for HCl absorption. Thus, the feasible chlorination temperature of MSCS should not exceed 750 °C according to the two possible reasons above mentioned. | ||
| Fig. 5 Effect of chlorination temperature on YHCl,N of cycled MSCS from various CO2 capture cycles (chlorination for 90 min in 0.20% HCl/N2 for balance). | ||
The chlorination temperature in the range of 700–800 °C has little effect on the cycled MSCS during the first 5 cycles. YHCl,10 at 700 °C and 800 °C is about 5% and 17% lower than that at 750 °C, respectively. However, as soon as the number of CO2 capture cycles exceeds 5, the chlorination temperature exhibits a great impact on the HCl capture capacity of the cycled MSCS. YHCl,20 at 700 °C and 800 °C is about 34% and 40% lower than that at 750 °C, respectively. This indicates that the number of CO2 capture cycles intensifies the effect of chlorination temperature on the HCl capture capacity of the cycled MSCS.
3.4 Effect of CO2 on HCl capture by cycled MSCS
When CO2 is present in the chlorination atmosphere, CO2 and HCl simultaneously react with calcium-based sorbents. Thus, carbonation has an impact on the chlorination of the sorbent. The effect of 10% CO2 in the chlorination atmosphere on the HCl removal capacity of the cycled MSCS is depicted in Fig. 6. In the presence of CO2, YHCl,0 of the MSCS is almost the same as YHCl,10, and both are lower than those in the absence of CO2, respectively. The volume fraction of CO2 is 50 times higher than that of HCl in the chlorination atmosphere, so the cycled MSCS may react with CO2 preferentially to form CaCO3. Subsequently, the formed CaCO3 reacts with HCl. However, the pore structure of CaCO3 is significantly less developed than that of CaO, so the HCl capture capacity of CaCO3 is lower. Therefore, CO2 shows an adverse impact on HCl capture by the cycled MSCS. Duo et al.55 also found that the direct chlorination property of CaCO3 is lower than that of CaO. YHCl,0 and YHCl,10 of MSCS in the presence of 10% CO2 is reduced by about 35% and 20% due to the presence of CO2, respectively. This means that the negative impact of CO2 on HCl capture by the cycled MSCS declines with an increasing number of CO2 capture cycles. This phenomenon can be explained as follows. In fact, the CO2 capture capacity of the calcium-based sorbent always decays with the number of CO2 capture cycles due to sintering. For example, our previous work47 has shown that the CO2 capture capacity of MSCS decreases by about 10% when increasing the cycle number from 1 to 10 in the absence of HCl. Therefore, when HCl is removed by the cycled MSCS, less CaCO3 is formed in the presence of CO2 with an increasing number of cycles. Accordingly, the negative impact of CO2 on the HCl removal of the cycled MSCS also becomes lower with an increasing number of cycles. | ||
| Fig. 6 Effect of CO2 on YHCl,N of cycled MSCS from various CO2 capture cycles (chlorination at 750 °C in 0.20% HCl/N2 for balance). | ||
3.5 Effect of HCl volume fraction on HCl capture by cycled MSCS
Fig. 7 demonstrates the effect of the HCl volume fraction on the HCl capture capacity of MSCS and carbide slag for 90 min of chlorination which has experienced various CO2 capture cycles. | ||
| Fig. 7 Effect of HCl volume fraction on YHCl,N of MSCS and carbide slag from various CO2 capture cycles (chlorination at 750 °C for 90 min). | ||
This shows that the HCl capture capacities of the cycled MSCS and the cycled carbide slag both almost increase linearly with HCl volume fraction. As the HCl volume fraction rises from 0.1% to 0.2%, YHCl,1 and YHCl,5 of MSCS increase by 150% and 170%, respectively. YHCl,1 of the carbide slag under 0.1%, 0.15% and 0.2% HCl are 78%, 45% and 9% higher than YHCl,5 under the corresponding HCl volume fraction, respectively. This reveals that the cycle number has a great effect on YHCl,N of the carbide slag under a low HCl volume fraction, but shows little effect under a high HCl volume fraction (e.g. 0.2% HCl). It is interesting to find that YHCl,1 and YHCl,5 of MSCS are almost equal, regardless of a high or low HCl volume fraction. This is possible because MSCS maintains a better pore structure with the aid of the support of MgO during the CO2 capture cycles, while the carbide slag suffered severe sintering and requires a high HCl volume fraction to offset the adverse effect of the change of pore structure. For the same HCl volume fraction and cycle number, MSCS exhibits higher YHCl,N than the carbide slag. YHCl,5 of MSCS under 0.15% and 0.2% HCl are approximately 2.1 and 1.7 times higher than those of the carbide slag, respectively.
3.6 Microstructure analysis
The SEM-EDS mapping of MSCS is illustrated in Fig. 8. The elements of Ca, O and Mg are evenly distributed on the surface of MSCS, which means that MgO is well mixed with CaO. The good dispersion of CaO and MgO in MSCS plays an important role in MgO as the support for CaO, and helps MSCS maintain a high sintering resistance during the CO2 capture cycles and a high HCl capture capacity. The EDS spectrogram of the MSCS in Fig. 8 is illustrated in Fig. 9. The mass ratio of Ca to Mg in Fig. 8 is approximately equal to 4.3:1, which agrees with that in MSCS.
 | ||
| Fig. 8 SEM-EDS mapping of MSCS (mass ratio of CaO:MgO = 80:20). | ||
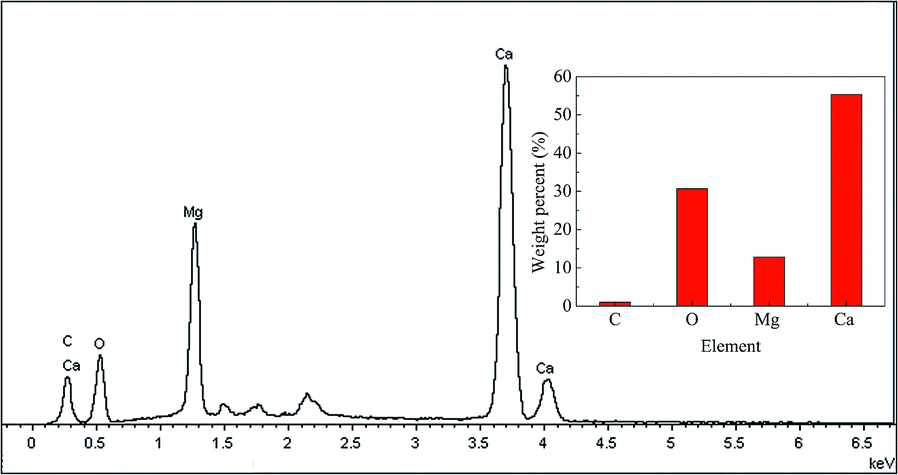 | ||
| Fig. 9 EDS spectrogram of MSCS in Fig. 8. | ||
The BET specific surface areas of MSCS and the carbide slag experiencing various CO2 capture cycles are shown in Table 2. The cycled MSCS displays a much larger specific surface area than the cycled carbide slag for the same CO2 capture cycles.
| Sample | Number of CO2 capture cycles | BET surface area (m2 g−1) |
|---|---|---|
| Carbide slag | 0 | 10.3 |
| Carbide slag | 10 | 4.2 |
| MSCS | 0 | 28.9 |
| MSCS | 10 | 20.8 |
The specific surface areas of MSCS after 0 and 10 CO2 capture cycles are 2.8 and 4.9 times higher than those of the carbide slag after the corresponding cycles, respectively. As the number of CO2 capture cycles rises from 0 to 10, the specific surface areas of MSCS and the carbide slag decrease by 28% and 59%, respectively. This suggests that MSCS can maintain a more stable microstructure due to the presence of MgO during repetitive CO2 capture cycles. During the combustion step in the preparation of MSCS, the by-product of biodiesel is rapidly burnt and the release of the gaseous combustion product as a foaming agent impacts the sorbent, which leads to the formation of the porous structure. Thus, the cycled MSCS from CO2 capture cycles shows a higher surface area. A larger specific surface area is beneficial for HCl capture by calcium-based sorbents.
The pore size distributions of MSCS and carbide slag after various CO2 capture cycles are plotted in Fig. 10. After the same cycles, the pore volume of MSCS is evidently larger than that of the carbide slag in the entire measured pore size range, especially the volume of the pores in the range of 2–10 nm. Xie et al.50 thought that the pores distributed in the 2–10 nm range were the most important areas for HCl absorption. The volume of pores in the range of 2–10 nm (V2–10 nm) and in the range of 10–150 nm range (V10–150 nm) of the two sorbents after 0 and 10 cycles are presented in Table 3. V2–10 nm of MSCS after 0 and 10 cycles are 4.5 and 5.7 times greater than that of the carbide slag after the same cycles, respectively.
 | ||
| Fig. 10 Pore size distributions of MSCS after various CO2 capture cycles (calcination at 850 °C for 10 min in pure N2 and carbonation at 700 °C for 20 min in 20% CO2/80% N2). | ||
| Samples | Number of CO2 capture cycles | V2–10 nm (cm3 g−1) | V10–150 nm (cm3 g−1) |
|---|---|---|---|
| a V2–10 nm denotes volume of pores in size range of 2–10 nm in diameter. V10–150 nm represents volume of pores in size range of 10–150 nm in diameter. | |||
| Carbide slag | 0 | 0.0035 | 0.0305 |
| Carbide slag | 10 | 0.0021 | 0.0185 |
| MSCS | 0 | 0.0159 | 0.1052 |
| MSCS | 10 | 0.0119 | 0.0661 |
Furthermore, V10–150 nm of MSCS after 0 and 10 cycles are 3.4 and 3.6 times greater than those of the carbide slag, respectively. The pores in the range of 10–150 nm in diameter are beneficial for HCl diffusion and absorption. The cycled MSCS from various CO2 capture cycles shows a higher HCl capture capacity, because it possesses better pore size distribution characteristics than the cycled carbide slag.
4 Conclusions
The cycled MSCS fabricated with industrial waste, i.e. carbide slag, magnesium nitrate hydrate and a by-product of biodiesel from transesterification by combustion, which had experienced carbonation/calcination cycles for CO2 capture was proposed to remove HCl. The reaction products of MSCS after HCl absorption are CaClOH, CaO and MgO. MgO in the MSCS is an inert support. MSCS which has not experienced CO2 capture cycles achieves a higher HCl capture capacity. The number of CO2 capture cycles exhibits little effect on the HCl capture capacity of the cycled carbide slag during 20 cycles. The cycled MSCS achieves a much higher HCl capture capacity than the cycled carbide slag for the same CO2 capture cycles. This may be attributed to the support of MgO in MSCS. Chlorination temperature in the range of 700–800 °C has little effect on the cycled MSCS during the first 5 cycles. However, as soon as the number of CO2 capture cycles exceeds 5, the chlorination temperature exhibits a great impact on the HCl capture capacity of the cycled MSCS. The optimum chlorination temperature of the cycled MSCS from CO2 capture cycles is 750 °C. CO2 shows an adverse impact on HCl capture by the cycled MSCS. The negative impact of CO2 on the HCl capture by the cycled MSCS declines with an increasing number of cycles. The good dispersion of CaO and MgO in MSCS plays an important role in MgO as the support for CaO, which helps MSCS maintain a high sintering resistance during the CO2 capture cycles and great HCl capture capacity. The cycled MSCS experiencing CO2 capture cycles possesses a higher surface area and better pore size distribution characteristics than the cycled carbide slag, so it exhibits a higher HCl capture capacity. It is proved that the highly active sorbent for CO2 capture from repetitive CO2 capture cycles can also capture HCl efficiently. Thus, MSCS is a promising synthetic sorbent for sequential CO2/HCl capture.Acknowledgements
Financial supports from the National Natural Science Foundation of China (51376003) and the Fundamental Research Funds of Shandong University, China (2014JC049) are gratefully appreciated. We thank Dr Hui Li for providing the by-product of biodiesel for this work.References
- D. Porbatzki, M. Stemmler and M. Müller, Biomass Bioenergy, 2011, 35(4), 397 Search PubMed.
- C. S. Chyang, Y. L. Han, L. W. Wu, H. P. Wan, H. T. Lee and Y. H. Chang, Waste Manage., 2010, 30(7), 1334 CrossRef CAS PubMed.
- M. Y. Wey, K. Y. Liu, W. J. Yu, C. L. Lin and F. Y. Chang, Waste Manage., 2008, 28(2), 406 CrossRef CAS PubMed.
- J. M. Yin and Z. S. Wu, Chin. J. Chem. Eng., 2009, 17(5), 849 CrossRef CAS.
- B. Shemwell, Y. A. Levendis and G. A. Simons, Chemosphere, 2001, 42(5–7), 785 CrossRef CAS PubMed.
- S. Fujita, N. Ogawa, T. Yamasaki, T. Fukuda, S. Sataka and K. Suzuki, Chem. Eng. J., 2004, 102(1), 99 CrossRef CAS.
- N. Verdone and P. D. Filippis, Chem. Eng. Sci., 2006, 61(22), 7487 CrossRef CAS.
- C. S. Chyang, Y. L. Han and Z. C. Zhong, Energy Fuels, 2009, 23, 3948 CrossRef CAS.
- T. Chin, R. Yan, D. T. Liang and J. H. Tay, Ind. Eng. Chem. Res., 2005, 44(10), 3742 CrossRef CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang and A. G. Russell, RSC Adv., 2013, 3(45), 22739 RSC.
- W. Z. Bi, T. Chen, R. Zhao, Z. Wang and J. H. Wu, RSC Adv., 2015, 5(44), 34913 RSC.
- E. J. Anthony, Greenhouse Gases: Sci. Technol., 2011, 1(1), 36 CrossRef CAS.
- H. C. Chen, C. S. Zhao and Q. Q. Ren, J. Environ. Manage., 2012, 93(1), 235 CrossRef CAS PubMed.
- V. Manovic and E. J. Anthony, Int. J. Environ. Res. Public Health, 2010, 7(8), 3129 CrossRef CAS PubMed.
- C. C. Dean, J. Blamey, N. H. Florin, M. J. Al-Jeboori and P. S. Fennell, Chem. Eng. Res. Des., 2012, 43(7), 836 Search PubMed.
- J. C. Abanades, E. J. Anthony, D. Y. Lu, C. Salvador and D. Alvarez, AIChE J., 2004, 50(7), 1614 CrossRef CAS.
- V. Manovic, J. P. Charland, J. Blamey, P. S. Fennell, D. Y. Lu and E. J. Anthony, Fuel, 2009, 88(10), 1893 CrossRef CAS.
- H. Ping and S. Wu, RSC Adv., 2015, 5(80), 65052 RSC.
- W. Liu, H. An, C. Qin, J. Yin, G. Wang and B. Feng, Energy Fuels, 2012, 26(5), 2751 CrossRef CAS.
- J. M. Valverde, J. Mater. Chem. A, 2012, 1(3), 447 RSC.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36(2), 260 CrossRef CAS.
- J. C. Abanades, B. Arias, A. Lyngfelt, T. Mattisson, D. E. Wiley and H. Li, Int. J. Greenhouse Gas Control, 2015, 40, 126 CrossRef CAS.
- A. Islam, S. H. Teo, E. S. Chan and T. Y. Yun, RSC Adv., 2014, 4(110), 65127 RSC.
- J. Sun, W. Q. Liu, Y. C. Hu, M. K. Li, X. W. Yang, Y. Zhang and M. H. Xu, Energy Fuels, 2015, 29(10), 6636 CrossRef CAS.
- Z. Li, N. S. Cai and Y. Huang, Ind. Eng. Chem. Res., 2006, 45(6), 1911 CrossRef CAS.
- M. Broda and C. R. Müller, Adv. Mater., 2012, 24(22), 3059 CrossRef CAS PubMed.
- L. Barelli, G. Bidini, A. D. Michele, F. Gallorini, C. Petrillo and F. Sacchetti, Appl. Energy, 2014, 127(7), 81 CrossRef CAS.
- A. Antzara, E. Heracleous and A. A. Lemonidou, Appl. Energy, 2015, 156(7), 331 CrossRef CAS.
- J. M. Valverde, A. Perejon and L. A. Perezmaqueda, Environ. Sci. Technol., 2012, 46(11), 6401 CrossRef CAS PubMed.
- X. Zhang, Z. Li, Y. Peng, W. Su, X. Sun and J. Li, Chem. Eng. J., 2014, 243(1), 297 CrossRef CAS.
- R. Y. Sun, Y. J. Li, H. L. Liu, S. M. Wu and C. M. Lu, Appl. Energy, 2012, 89(1), 368 CrossRef CAS.
- S. F. Wu and Y. Q. Zhu, Ind. Eng. Chem. Res., 2010, 49(6), 2701 CrossRef CAS.
- Y. C. Hu, W. Q. Liu, J. Sun, M. K. Li, X. W. Yang, Y. Zhang and M. H. Xu, Chem. Eng. J., 2015, 273(8), 333 CrossRef CAS.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2009, 48(19), 8906 CrossRef CAS.
- V. Manovic, P. S. Fennell, J. A. Moham and E. J. Anthony, Ind. Eng. Chem. Res., 2013, 52(23), 7677 CrossRef CAS.
- V. Manovic, Y. Wu and E. J. Anthony, Environ. Sci. Technol., 2012, 46(22), 12720 CrossRef CAS PubMed.
- V. Manovic and E. J. Anthony, Fuel, 2011, 90(1), 233 CrossRef CAS.
- H. C. Chen, C. S. Zhao, Y. M. Yang and P. P. Zhang, Appl. Energy, 2012, 91(1), 334 CrossRef CAS.
- H. C. Chen, C. S. Zhao and W. W. Yu, Appl. Energy, 2013, 112(4), 67 CrossRef CAS.
- A. Silabna, M. Narcida and D. P. Harrison, J. Exp. Biol., 1996, 146(1), 149 Search PubMed.
- J. M. Valverde, P. E. Sanchez-Jimenez and L. A. Perez-Maqueda, Appl. Energy, 2015, 138(C), 202 CrossRef CAS.
- Y. Q. Qiao, J. Y. Wang, L. Huang, Q. W. Zheng, D. O'Hare and Q. Wang, RSC Adv., 2015, 5(101), 82777 RSC.
- B. Marcin, M. K. Agnieszka and R. M. Christoph, Adv. Funct. Mater., 2014, 24(36), 5753 CrossRef.
- C. Luo, Y. Zheng, Y. Q. Xu, N. Ding, Q. W. Shen and C. G. Zheng, Chem. Eng. J., 2015, 267(5), 111 CrossRef CAS.
- W. Q. Liu, B. Feng, Y. Q. Wu, G. X. Wang, J. Barry and C. J. Da, Environ. Sci. Technol., 2010, 44(8), 3093 CrossRef CAS PubMed.
- W. Q. Liu, J. J. Yin, C. L. Qin, B. Feng and M. H. Xu, Environ. Sci. Technol., 2012, 46(20), 11267 CrossRef CAS PubMed.
- X. T. Ma, Y. J. Li, L. Shi, Z. R. He and Z. Y. Wang, Appl. Energy, 2016, 168(4), 85 CrossRef CAS.
- Y. J. Li, R. Y. Sun, C. T. Liu, H. L. Liu and C. M. Lu, Int. J. Greenhouse Gas Control, 2012, 9(3), 117 CrossRef.
- X. Xie, Y. J. Li, W. J. Wang and L. Shi, Appl. Energy, 2014, 135(12), 391 CrossRef CAS.
- X. Xie, Y. J. Li, C. T. Liu and W. J. Wang, Fuel Process. Technol., 2015, 138(10), 500 CrossRef CAS.
- H. Li, S. L. Niu, C. M. Lu, M. Q. Liu and M. J. Huo, Sci. China: Technol. Sci., 2014, 57(2), 438 CrossRef CAS.
- J. Partanen, P. Backman, R. Backman and M. Hupa, Fuel, 2005, 84, 1674 CAS.
- M. Daoudi and J. Walters, Chem. Eng. J., 1991, 47(1), 11 CrossRef CAS.
- C. E. Weinell, P. I. Jensen and K. Dam-Johansen, Ind. Eng. Chem. Res., 1992, 31(1), 164 CrossRef CAS.
- W. Duo, J. P. K. Sevill, N. F. Kirkby and R. Clift, Chem. Eng. Sci., 1994, 49(24), 4429 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |