
Effects of paclitaxel (PTX) prodrug-based self-assembly peptide hydrogels combined with suberoylanilide hydroxamic acid (SAHA) for PTX-resistant cancer and synergistic antitumor therapy†
 *a
*a
Abstract
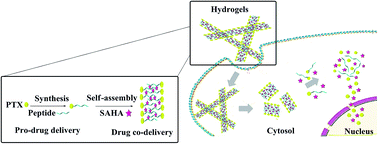
In this work, we designed a self-assembly peptide hydrogel encapsulated paclitaxel (PTX) and suberoylanilide hydroxamic acid (SAHA) for cancer co-delivery (Nap–PTX–SAHA). The hydrogels exhibited uniform nanofibers that entangle to form stable networks and pore microstructures (about 140–180 nm) resulting in absorption and storage of hydrophobic drugs. The circular dichroism (CD) spectra of hydrogels exhibit β-turn-like features. Nap–PTX–SAHA hydrogels achieved control over different types of drugs loading and released by diffusion and relaxation processes. Hydrogels exhibited better therapeutic effect and greater synergetic effect compared to the free drugs in vitro and in vivo. Injection of the hydrogels appears to achieve long-term drug release with higher efficacy and better biocompatibility than free drugs. As for the drug biodistribution studies in tumor-bearing mice, the PTX and SAHA from Nap–PTX–SAHA gels uptake in liver, lung and tumor were more than that of free PTX and SAHA. In comparison with free PTX or SAHA, the Nap–PTX–SAHA gels lower the uptake of released drug significantly in the heart, spleen, and kidney to reduce the cardiotoxicity and renaltoxicity. In conclusion, the Nap–PTX–SAHA gels certainly reduce the side effects of PTX and SAHA, enhance synergistic anticancer effects and suppress the drug resistance of PTX. This strategy can be a basis for designing appropriate clinical trials and will hold promise in nanomedicine for different drug combinations.
 Please wait while we load your content...
Please wait while we load your content...

