
Synthesis and optoelectronics properties of diblock copolymer of P3HT containing thiol-side chains and its hybrid nanocomposite†
Yisha Qiao,
Yixuan Du,
Yinfeng Liu and
Yunbo Li*
School of Materials Science & Engineering, Shanghai University, Shanghai 200072, China. E-mail: ybli@shu.edu.cn; liyunbo@shu.edu.cn
First published on 16th November 2016
Abstract
Although the solid-state microstructure of semiconducting polymers is well known to influence properties in optoelectronic devices, the control of desired aggregation in solution and film remains relatively rare. Here, diblock copolymer poly(3-hexylthiophene)-b-poly(3-thiophenehexanethiol) (DP-P3HT-SH) has been synthesized by a simple synthetic technique, and different aggregation states of DP-P3HT-SH and its respective hybrid nanostructures DP-P3HT-S-AuNPs had been prepared to research the influence of aggregate morphology on photoelectric performance. The diblock copolymer DP-P3HT-SH could be prepared with different aggregation states, i.e., global (G), leaf-like (L) and elliptical (E) shape states. The DP-P3HT-S-AuNPs composites exhibited a wider absorption band than the DP-P3HT-SH polymer and showed photoluminescence (PL) quenching. The amount of PL quenching of the three aggregation states was 23%, 11% and 21%, respectively, which illustrated that the charge transfer between GDP-P3HT-SH and AuNPs was the most efficient among these three aggregation states. In addition, the conductivity of these three aggregation states of DP-P3HT-SH could be improved by the addition of AuNPs and the increase of film thickness. Therefore, the semiconducting polymer–metal composites could be used to design new optoelectronics materials by tuning the aggregation state of the diblock copolymer to modulate device characteristics.
Introduction
Polythiophene and its derivatives are important semiconducting polymers with optoelectronic properties, low cost and good processability.1 The properties of polythiophene derivatives are determined by their structure, regularity and self-aggregated morphology.2 As one of the key factors, the aggregate morphology has been studied in semiconducting materials. R. D. McCullough has reported that the introduction of alkyl to polythiophene contributes to the polymer self-assembly into a nanometer fiber structure which improved the efficiency of the photoelectronic device.3 X. Yu and co-works reported that different self-assembled morphologies of spheres, lamellae, worm nanofibers and crystalline nanoribbons of conjugated block copolymers polystyrene-b-poly(3-hexylthiophene) (PS-b-P3HT) effected field-effect mobility.4 B. A. G. Hammer prepared nanowires and cross-linked nanowires structures of P3HT-b-poly(3-(3-thioacetylpropyl)oxymethylthiophene) (P3TT) which leads to different transfer and output characteristics of field-effect transistor.5 Seth B. Darling group reported that block copolymer self-assembly can be utilized to rationally design and control the shape and dimension of resulting nanostructures,6 such as the application of the self-assembled P3HT-b-PLLA in organic or hybrid solar energy devices.7 Verduzco, R. group reported that conjugated diblock copolymers may be useful for achieving active layers in organic photovoltaics and light-emitting diodes, and the important application of supramolecular conjugated block copolymers for improving the performance of all-polymer organic photovoltaics.8 Among the polythiophene derivatives materials, regioregular poly(3-hexylthiophene) (P3HT) has shown the good performance in electricity devices.9 The excellent charge-transport properties of P3HT have been attributed to microstructure of semiconductor. The solid-state microstructure of semiconducting polymers is known to affect properties relevant for their function in optoelectronic devices.10In addition, large aggregate structures conjugated diblock copolymers are valued in photovoltaic device in recent years. Organic photovoltaics (OPVs) device performance depends critically on the polymer morphology of the active layer, which ideally promotes efficient exciton formation, charge dissociation, and charge transport to the respective electrodes.2b,8b For example, Ryan C. Hayward group had prepared different morphologies of perylene diimide (PDI) fibers with width of 150–500 nm or 70–200 nm.11 Todd Emrick and his coworkers prepared large size polymer P3HT-b-P3MT fibrils with width 15–25 nm and length 0.5–5.0 μm.2b Accordingly, it is necessary to study the influence of large structures with different morphologies on the photoelectronic performance of organic semiconductor materials.
In recent years, semiconducting polymer–inorganic composites have attracted increasing interest for applications in organic optoelectronics devices mainly due to their optical and electricity properties.12 In particular, conjugated diblock copolymers P3HT have already been used in electronic devices.13 Simultaneously, many works have been done to design hybrid nanostructures based on conjugated polymers bond with inorganic like CdSe14 and ZnO,15 and form semiconductors. Jih-Jen Wu and his colleagues demonstrated that the introduction of Au@silica NPs could enrich 30% of short-circuit current density in P3HT solar cell.16 Solar cells fabricated using TiO2-coated ZnO NRs and P3HT showed efficiencies of up to 0.76%, and a 58% improvement over TiO2-free equivalent devices.17 Accordingly, the inorganic had important influence on photoelectronic performance of semiconducting materials.
Among them, semiconducting polymer–metal hybrid system has been paid attention due to exciton plasmon interactions which are different from its individual counterpart.12a This hybrid system exhibits enhanced radiative rates and exciton plasmon energy transfer due to weak coupling regimes.18 The semiconducting polymer–metal nanoparticles could provide appropriate electron donor and electron acceptor which would produce electric dipole in the composite and form the stable electronic channels. The energy transfer/charge separation could occur when the semiconducting polymer–metal nanoparticles were excited. C. Fan had described the high efficiency energy transfer from the conjugated polymer to gold NPs.19 V. Ruiz had reported that the addition of gold nanoparticles could significantly improve the conductivity of ultrathin poly(3-hexylthiophene)/gold nanoparticle blend films.12f
In this study DP-P3HT-SH has been synthesized by the simple solution based synthetic technique and three different aggregation states DP-P3HT-SH and their respective composites DP-P3HT-S-AuNPs hybrid nanostructures have been prepared. Absorption and photoluminescence spectra have been studied. In addition, the conductivity of P3HT-S-AuNPs films made with three different aggregation states were systematically investigated.
Experimental section
Chemicals and materials
Tetrahydrofuran (THF, from Sinopharm) was freshly distilled before use. [1,3-Bis(diphenylphosphino)propane]-dichloronickel(II) (Ni(dppp)Cl2), 3-bromothiophene (97%), 2,5-dibromo-3-hexylthiophene (99%), N-bromosuccinimide (NBS, 99%), magnesium turnings and chloroform (CHCl3) were purchased from Sinopharm and used as-received. Isopropylmagnesium chloride (2.0 M solution in THF) and 4-methoxyphenol were purchased from Aldrich Chemicals. 2-Thiourea was purchased from TCL. 1,6-Dibromohexane was purchased from Alfa Aesar. Gold(III) chloride trihydrate (HAuCl4) and trisodium citrate were obtained from Sinopharm.Synthesis of 3-[6-(4-methoxyphenoxy)hexyl]thiophene
4-Methoxyphenol (3.1 g, 0.025 mol), KOH (1.2 g, 0.021 mol), and 20 ml of dried methyl alcohol was added into a three-necked 500 ml round-bottom flask with a magnetic stirring bar. The solution was stirred for 1.0 hour. 1,6-Dibromohexane (10.0 g, 0.041 mol) was added into the reaction solution and the mixture was refluxed overnight. The mixture then was filtered to remove the KBr. Methanol was evaporated and the mixture was redissolved in hexane. 2.0 mol L−1 NaOH (aq), 1.0 mol L−1 NaCl (aq) and water were prepared to wash the mixture sequentially to achieve a neutral state. Under the condition of low pressure, the unreacted 1,6-dibromohexane was removed by distillation. The residue was redissolved in hexane, and the product was filtered after the solution was cooled down for recrystallization. The product of 1-(6-bromohexyl)-4-methoxybenzene was obtained.20 1-(6-Bromohexyl)-4-methoxybenzene (3.01 g, 0.01 mol) was added into 20 ml of anhydrous ether firstly and magnesium turnings (0.21 g, 0.009 mol) were added under inert atmosphere. The mixture was then refluxed for 2.0 hours. 1.0 ml 3-bromothiophene and Ni(dppp)Cl2 (10.0 mg, 0.00002 mol) at 0 °C were added into the mixture. The mixture subsequently was refluxed for 2.0 hours. 10.0 ml of 1.0 mol L−1 HCl and 10.0 ml distilled water was added into hydrolysis reaction system. The system was extracted with ether, washed to neutrality and dried with the combined organic phases. A light yellow product was obtained after evaporate the solvent.21 1H NMR (500 MHz, CDCl3): 7.26 (s, 1H), 6.99 (t, 2H), 6.81 (s, 1H), 2.81 (m, 2H), 2.59 (m, 3H), 1.74 (m, 2H), 1.53–1.33 (m, 4H) (ESI, Fig. S1†) (Scheme 1). | ||
| Scheme 1 Schematic illustration for the synthesis of the 3-[6-(4-methoxyphenoxy)hexyl]thiophene. | ||
Synthesis of 2,5-dibromo-3-(6-bromohexylthiophene)
3-[6-(4-Methoxyphenoxy)hexyl]thiophene (2.1 g, 0.007 mol) was added into a three-necked 100 ml round-bottom flask with a magnetic stirring bar under inert atmosphere. A mixture of HBr and acetic anhydride (6 ml![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ml) was added into the flask. The reaction mixture was heated at 100 °C to reflux for 24 hours. The mixture was extracted with ether and the solution was washed to neutrality with saturated aqueous NaHCO3 solution. The solution was transferred into a silica gel column, and eluted by hexane. A colorless oil product 3-(6-bromohexylthiophene) was obtained after removing the solvent.21 3-Bromohexylthiophene (1.5 g, 0.006 mol) was added into a 100 ml round-bottom flask for stirring, then the THF and acetic acid (6 ml:6 ml) mixed liquid was added into the flask. n-Bromosuccinimide (2.0 g, 0.011 mol) was added into the compound and heated at 30 °C for an hour. Terminating reaction was carried out by distilled water (10 ml), then the solution was extracted with diethyl ether (Et2O) (30 ml). The organic extracts were washed by distilled water (30 ml) and saturated NaHCO3 solution (30 ml); then anhydrous MgSO4 was used to dry the organic layer. After evaporating the solution, the product was transferred into a silica gel column and eluted with petroleum ether. The final colorless product was 2,5-dibromo-3-(6-bromohexylthiophene).22 1H NMR (500 MHz, CDCl3): 7.26 (s, 1H), 6.99 (t, 2H), 3.77 (s, 1H), 3.67 (t, 2H), 2.31 (t, 2H), 1.87 (m, 2H), 1.25–1.53 (m, 6H) (ESI, Fig. S2†) (Scheme 2).
:6 ml) was added into the flask. The reaction mixture was heated at 100 °C to reflux for 24 hours. The mixture was extracted with ether and the solution was washed to neutrality with saturated aqueous NaHCO3 solution. The solution was transferred into a silica gel column, and eluted by hexane. A colorless oil product 3-(6-bromohexylthiophene) was obtained after removing the solvent.21 3-Bromohexylthiophene (1.5 g, 0.006 mol) was added into a 100 ml round-bottom flask for stirring, then the THF and acetic acid (6 ml:6 ml) mixed liquid was added into the flask. n-Bromosuccinimide (2.0 g, 0.011 mol) was added into the compound and heated at 30 °C for an hour. Terminating reaction was carried out by distilled water (10 ml), then the solution was extracted with diethyl ether (Et2O) (30 ml). The organic extracts were washed by distilled water (30 ml) and saturated NaHCO3 solution (30 ml); then anhydrous MgSO4 was used to dry the organic layer. After evaporating the solution, the product was transferred into a silica gel column and eluted with petroleum ether. The final colorless product was 2,5-dibromo-3-(6-bromohexylthiophene).22 1H NMR (500 MHz, CDCl3): 7.26 (s, 1H), 6.99 (t, 2H), 3.77 (s, 1H), 3.67 (t, 2H), 2.31 (t, 2H), 1.87 (m, 2H), 1.25–1.53 (m, 6H) (ESI, Fig. S2†) (Scheme 2).
 | ||
| Scheme 2 Schematic illustration for the synthesis of the 3-(6-bromohexylthiophene) and 2,5-dibromo-3-(6-bromohexylthio-phene). | ||
Synthesis of diblock copolymer poly(3-hexylthiophene)-b-poly(3-(6-bromohexylthiophene)) (P3HT-b-P3BPT)
2,5-Dibromo-3-hexylthiophene (1.63 g, 0.005 mol) and 2,5-dibromo-3-(6-bromohexylthiophene) (0.41 g, 0.001 mol) were separately dissolved in THF (10 ml mg−1) in a 100 ml round-bottom flask. 2.0 mol L−1 isopropylmagnesium chloride (0.58 g, 0.005 mol) and 2.0 mol L−1 isopropylmagnesium chloride (0.116 g, 0.0011 mol) were separately added into the compound sequentially and refluxed for 2 hours. 1,3-Bis(diphenylphosphino)propane]-dichloronickel catalyst (15.0 mg, 0.00003 mol) was added into the flask reacted with the 2,5-dibromo-3-hexylthiophene Grignard for 1 hour. Then the 2,5-dibromo-3-(6-bromohexylthiophene) Grignard solutions was added into the above reaction system and reacted for another 1 hour. 10 ml methanol was used to quench the polymerization. The polymer was precipitated in methanol and centrifuged, then the solvent was decanted off and the polymer was collected. The polymer was purified by sequential Soxhlet extractions using methanol, hexane, and chloroform. The chloroform fraction was dried to obtain the final polymer as a deep purple solid.5 1H NMR (500 MHz, CDCl3): 6.98 (s, 1H), 3.71 (t, 2H), 2.82 (t, 2H), 1.73 (m, 2H), 1.50 (m, 4H) (ESI, Fig. S3†) (Scheme 3). | ||
| Scheme 3 Schematic illustration for the synthesis of the poly(3-hexylthiophene)-b-poly(3-(6-bromohexylthiophene)). | ||
Synthesis of diblock copolymer poly(3-hexylthiophene)-b-poly(3-(6-thiolhexylthiophene)) (DP-P3HT-SH)
0.91 g P3HT-b-P3BPT and 2-thiourea (0.15 g, 0.002 mol) was dissolved in ethanol (20 ml, 95%) in a round-bottom flask and refluxed for 2 hours. KOH solution (10 ml, 0.3 mol L−1) was added into the compound and refluxed for 4 hours; then 0.05 ml H2SO4 (98%) in 5 ml distilled water was added in the reaction system to neutralize excess alkali. The solution was extracted with hexane and washed by distilled water, and then the anhydrous MgSO4 was used to dry the organic layer. The resulting polymer was purified by sequential Soxhlet extractions using methanol, hexane, and chloroform. The chloroform fraction was dried and the obtained polymer is a deep purple solid.5 1H NMR (500 MHz, CDCl3): 7.00 (s, 1H), 2.70 (t, 2H), 2.59 (m, 2H), 1.73 (m, 2H), 1.25–1.70 (m, 8H), 0.85 (t, 3H) (ESI, Fig. S4†) (Scheme 4). | ||
| Scheme 4 Schematic illustration for the synthesis of the poly(3-hexylthiophene)-b-poly(3-(6-thiolhexylthiophene)). | ||
Preparation of different aggregate states DP-P3HT-SH in solution
Global state DP-P3HT-SH (GDP-P3HT-SH) was prepared as followed: DP-P3HT-SH was dissolved in THF and kept concentration of 1.0 mg ml−1 at 20 °C; then the copolymer was stirred vigorously for 20 hours at 25 °C and ultrasonic 5 min. Leaflike state DP-P3HT-SH (LDP-P3HT-SH) was obtained by the following method: DP-P3HT-SH solution in THF was heated to 80 °C for 30 min; then the solution was cooled to room temperature naturally overnight. Elliptical shape DP-P3HT-SH (EDP-P3HT-SH) was prepared by heating the LDP-P3HT-SH solution to 50 °C for 12 hours; then the polymer was cooled to 40 °C for 48 hours.Synthesis of gold nanoparticles (AuNPs)
The gold nanoparticles in water were synthesized as reported by G. Frens.23 Aqueous solution of HAuCl4 (1 × 10−4 g ml−1, 50 ml) was boiled under gentle stirring. Then trisodium citrate (0.01 g ml−1, 0.5 ml) was added into the HAuCl4 aqueous solution with stirring. After 30 second the mixed solution turned faintly blue and 2 min later the blue colour suddenly changed into a brilliant red indicating the formation of monodisperse spherical particles. After further vigorous stirring for 5 min, the product was collected. Next, 10 ml of hexane 10 mg of sodium oleate were added into 10 g of the product in water prepared above. The mixture was emulsified by vigorous stirring at room temperature for 2 hours. Magnesium chloride (0.12 g, 0.001 mol) in 1.5 ml of water was then added with stirring. The mixture was transformed from an emulsion into two liquid phases after 4 h sedimentation. The hexane phase containing AuNPs was collected. A molecular sieve was used to remove the residual water.24Preparation of the hybrid particles DP-P3HT-S-AuNPs
The hybrid particles DP-P3HT-S-AuNPs were obtained as followed: gold nanoparticles in hexane and DP-P3HT-SH in THF were stirred for 6 hours. The hybrid particles were purified by centrifugation. Next the precipitated was dissolved in THF. This purification procedure was repeated for another two times. The product was dispersed in THF finally and the concentration of DP-P3HT-S-AuNPs solution was kept at 1.0 mg ml−1.Characterization techniques
Transmission electron microscopy (TEM) images were obtained by a JEOL 200CX electron microscope equipped with a Model GATAN782 CCD camera at an operating voltage of 120 kV. The samples for TEM studies were prepared by placing one droplet of the sample deposited onto carbon-coated copper grids. An Agilent 8453 UV-vis spectrophotometer (Agilent technologies, CA, USA) is utilized to analyze the UV-vis absorption property of the sample. The slit width was 1 nm during the measurements. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet 380 FTIR spectrophotometer. The photoluminescence (PL) experiments were carried out on a RF5301 fluorescence spectrophotometer for the films and solution. The exciting wavelength and slit width are at 470 nm and 5 nm, respectively. The film thickness was measured by a Surfcorder ET 150 profiler. All electric conductivity measurements were made along the film plane using a VC890C+ multimeter. The samples were prepared as followed: different aggregation states for the diblock copolymer P3HT-SH (DP-P3HT-SH) at 1.0 mg ml−1 in THF with different volume gold nanoparticles in hexane. Several fluorine-doped tin oxide (FTO) were prepared and cleaned for further works. The composites DP-P3HT-S-AuNPs and DP-P3HT-SH solutions were dropped onto precleaned FTO glass to get a uniform film, and the film thickness was controlled in the range from 0.7 μm to 2.4 μm by adjusting the dropping layer. The substrate of the sample films for conductivity measurement was the etched FTO glass with a channel 1 mm wide. 2-Probe methods using a VC890C+ multimeter were employed for conductivity measurement in air. The conductivity was calculated from Ohm's law:25| σ = Iw/Vdl | (1) |
Results and discussion
Size distribution of gold nanoparticles and FTIR spectra of P3HT-b-P36THT, P3HT-b-P36THT/AuNPs
Fig. 1a showed the size distribution of gold nanoparticles in hexane. The average particle diameter of gold nanoparticles is about 22.5 nm. The smaller of the size of gold nanoparticles contain less number atoms which could make nanometer system appeared many new characteristics different from conventional materials. The gold nanoparticles were used in the next work.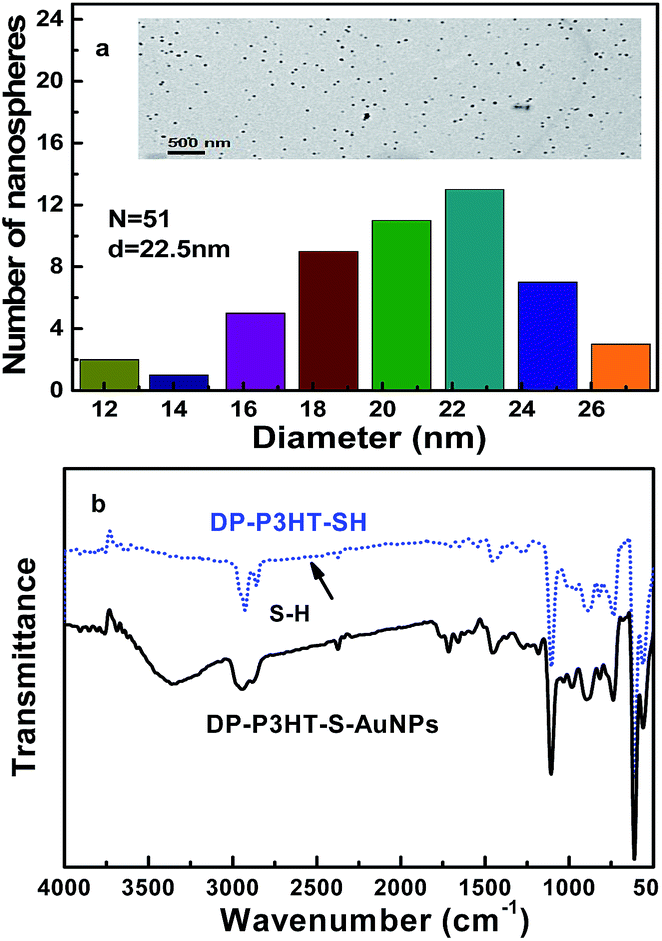 | ||
| Fig. 1 Size distribution of gold nanoparticles (AuNPs) in hexane (the inset shows the transmission electron morphology images of gold nanoparticles) (a) and FTIR spectra of the DP-P3HT-SH and DP-P3HTS-AuNP hybrid particles (b). | ||
Fig. 1b showed the FTIR spectra of the DP-P3HT-SH and DP-P3HT-S-AuNPs hybrid particles. The polymer chain included the following main characteristic absorption bands: 2938 cm−1 bands of the C–H stretching mode of thiophene ring; 2852 cm−1 bands of the C–H stretching mode outside of the thiophene ring side chain; 1701 cm−1, 1538 cm−1 and 1465 cm−1 bands of symmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching; 1315 cm−1 bands of the –CH3 stretching mode outside of the thiophene ring side chain; 1118 cm−1 bands of C–Br stretching mode of thiophene ring; 823 cm−1 bands of CH out of plane deformation; 723 cm−1, 655 cm−1 bands of C–S–C stretching mode of thiophene ring. In addition, the weak peak at about 2565 cm−1 was the bands of the S–H stretching mode outside of the thiophene ring side chain.26 Therefore the resulting product was confirmed that DP-P3HT-SH was the successful synthesized. The IR spectra of the DP-P3HT-S-AuNPs hybrid particles and of DP-P3HT-SH are similar which indicates that thiol is indeed part of the composite. What's more, the intensity of absorption peak of the obtained product becomes lower compared with that of DP-P3HT-SH, which indicates that the DP-P3HT-SH polymer was thiolated gold nanoparticles.27
C stretching; 1315 cm−1 bands of the –CH3 stretching mode outside of the thiophene ring side chain; 1118 cm−1 bands of C–Br stretching mode of thiophene ring; 823 cm−1 bands of CH out of plane deformation; 723 cm−1, 655 cm−1 bands of C–S–C stretching mode of thiophene ring. In addition, the weak peak at about 2565 cm−1 was the bands of the S–H stretching mode outside of the thiophene ring side chain.26 Therefore the resulting product was confirmed that DP-P3HT-SH was the successful synthesized. The IR spectra of the DP-P3HT-S-AuNPs hybrid particles and of DP-P3HT-SH are similar which indicates that thiol is indeed part of the composite. What's more, the intensity of absorption peak of the obtained product becomes lower compared with that of DP-P3HT-SH, which indicates that the DP-P3HT-SH polymer was thiolated gold nanoparticles.27
1H NMR spectra of the thiol-functionalized diblock copolymers (DP-P3HT-SH)
Fig. 2 showed the 1H NMR spectrum which was used to characterize the synthesized DP-P3HT-SH. The final product had eight main bonds of carbon–hydrogen which corresponded to peaks in 1H NMR spectrum. 1H NMR (500 MHz, CDCl3): 7.00 (s, 1H), 2.70 (t, 2H), 2.61 (m, 2H), 1.73 (m, 2H), 1.25–1.70 (m, 8H), 0.85 (t, 3H). The appearance of the methylene proton peaks next to thiol group at 2.61 ppm could illustrate the existence of S–H bonds. The 1H NMR spectrum was consistent with the studies reported by G. Konstantatos group who had synthesized the thiol-functionalized block copolymer P3HT-SH.1c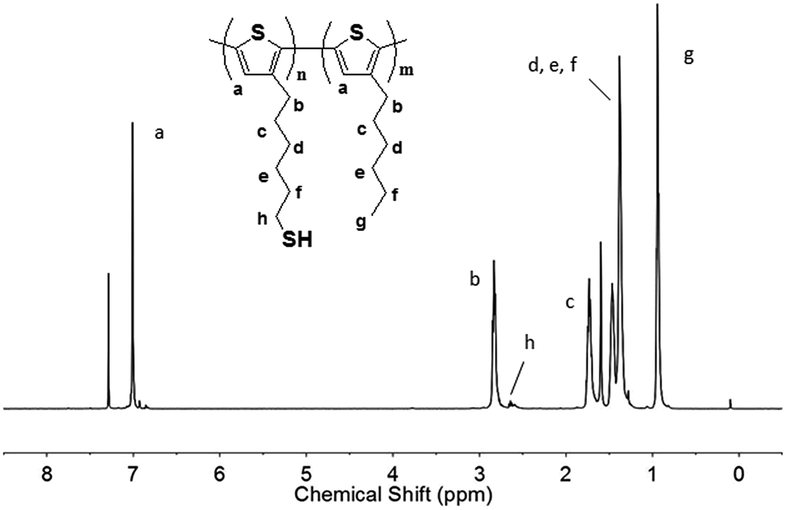 | ||
| Fig. 2 1H NMR spectra of poly(3-hexylthiophene)-b-poly(3-(6-thiolhexylthiophene)) (DP-P3HT-SH) in CDCl3. | ||
Morphology of thiol-functionalized diblock copolymer DP-P3HT-SH and its hybrid nanoparticles
TEM analysis was used to observe the nanomorphology of DP-P3HT-SH and their respective composites DP-P3HT-S-AuNPs. Fig. 3A and B showed images of GDP-P3HT-SH and hybrid particles GDP-P3HT-S-AuNPs. DP-P3HT-SH self-aggregated into global nanoparticles with average diameter of about 260 nm. These nanoparticles were uniform and stable. The gold nanoparticles were fixed on the surface of GDP-P3HT-SH due to the side-chain thiol leaved outside of the polymer through Au–S band and formed GDP-P3HT-S-AuNPs hybrid particles.28 The GDP-P3HT-SH polymer and its hybrid particles GDP-P3HT-S-AuNPs were investigated in next work. | ||
| Fig. 3 TEM images of different aggregation states DP-P3HT-SH (A, C and E) and P3HT-S-AuNPs hybrid particles (B and D) (inset shows image of DP-P3HT-SH in THF without operation). | ||
Fig. 3C and D showed images of LDP-P3HT-SH and hybrid particles LDP-P3HT-S-AuNPs. The lengths of LDP-P3HT-SH were observed to vary from ∼700 to 1500 nm, with widths ranging from 200 to 300 nm. It appeared gaps in LDP-P3HT-SH polymer. This morphology polymer induced the gold nanoparticles combining with LDP-P3HT-SH (shown in Fig. 3D) and formed LDP-P3HT-S-AuNPs hybrid particles. It could be observed that gold nanoparticles were well dispersed in LDP-P3HT-SH, revealing that the combination between LDP-P3HT-SH and AuNPs could prevent the aggregation of gold nanoparticles. The LDP-P3HT-SH and its composite were researched in the following work.
The elliptical shape state of diblock copolymer P3HT-SH (EDP-P3HT-SH) could be seen in Fig. 3E. It was prepared on the basis of LDP-P3HT-SH and the EDP-P3HT-SH had a steady morphology. The widths and lengths of elliptical-like polymer particles were ranging from 1.5 μm to 2.5 μm and from 2.5 μm to 4.0 μm, respectively. It had no image of EDP-P3HT-S-AuNPs because gold nanoparticles couldn't be observed clearly. EDP-P3HT-SH and EDP-P3HT-S-AuNPs were observed by TEM and kept the same morphology after several days. Therefore EDP-P3HT-SH and EDP-P3HT-S-AuNPs were chosen to use in the following study.
Absorption of three different aggregation states DP-P3HT-SH film and solution
The solid state normalized UV-visible absorption spectra of different aggregation states of diblock copolymer P3HT-SH film and DP-P3HT-S-AuNPs composites film were shown in Fig. 4a and b. In addition, the spectra of DP-P3HT-SH compared with respective P3HT-S-AuNPs composites could be observed in Fig. 4c–e. In Fig. 4a, three aggregation states of DP-P3HT-SH exhibited broad absorption spectra between 300 nm to 800 nm including a main absorption peak at around 510 nm and the two vibronic “shoulders” at 553 nm and 605 nm. The absorption peak centered at 510 nm was attributed to the π–π* transition. The peak centered at 553 nm was due to the absorption of extended conjugation of DP-P3HT-SH in the solid film and the absorption peak at 605 nm was caused by the interchain stacking of DP-P3HT-SH.29 Compared with other two aggregation states of DP-P3HT-SH, the GDP-P3HT-SH appeared a red shift by 11 nm that maybe arise from relatively relaxed and ordered conformations.12a As can be seen from Fig. 3b, the absorption spectra of DP-P3HT-S-AuNPs composites were listed. Three peaks at 515 nm, 559 nm and 609 nm were observed. The DP-P3HT-S-AuNPs composites showed a slight larger absorption band and stronger absorption peak compared with corresponding DP-P3HT-SH, respectively. It could be due to the further increased conjugation of main chain and the stronger interaction between gold nanoparticles and DP-P3HT-SH.15b Therefore, the DP-P3HT-S-AuNPs composites could absorb more visible light than DP-P3HT-SH, respectively. In addition, as the thickness is kept same for all these films, the change in the absorption spectra is believed to be due to inter-chain interaction of different aggregation states and the addition of gold nanoparticles, and not because of different film thickness.29 | ||
| Fig. 4 Solid state normalized UV-visible absorption spectra of different aggregation states DP-P3HT-SH (a) (the inset shows the plasmon band of pure AuNPs), respective composites DP-P3HT-S-AuNPs hybrid particles (b) and respective DP-P3HT-SH comparing with P3HT-S-AuNP composites films (c–e). | ||
The normalization absorption spectra of DP-P3HT-SH films and DP-P3HT-S-AuNPs composites films were compared in Fig. 4c–e. Comparing with DP-P3HT-SH, DP-P3HT-S-AuNPs composites appeared a red shifting by 7 nm, 10 nm and 9 nm at the maximum absorption peaks, respectively. This red shifting was attributed to the distributed of AuNPs on the surface of DP-P3HT-SH by Au–S chemical bonds.
Fig. 5 showed the UV-vis absorption spectra of three different aggregation states DP-P3HT-SH solution and its respective DP-P3HT-S-AuNPs hybrid nanoparticles in THF. For the spectrum of DP-P3HT-SH solution, they showed an absorption band between 350 nm and 650 nm which was narrower than corresponding film varying from 300 nm to 800 nm in Fig. 4a. Compared the absorption band of different aggregation states DP-P3HT-SH solution with its solid films, it can be concluded that the DP-P3HT-SH solid state were better to absorb sunlight due to its absorption spectrum covered the visible light region. For the spectra of DP-P3HT-SH solution, three peaks located at around 505 nm, 543 nm and 597 nm which corresponded to 510 nm, 553 nm and 605 nm in the solid film, respectively. That is to say DP-P3HT-SH solid film appeared a red shifting to its solution state. Compared to DP-P3HT-SH, the absorption intensity of composites DP-P3HT-S-AuNPs presented a certain degree increase due to the addition of AuNPs.
 | ||
| Fig. 5 UV-visible absorption spectra of different aggregation states DP-P3HT-SH solution (a) and its respective DP-P3HT-S-AuNPs hybrid particles solution (b). | ||
Photoluminescence of DP-P3HT-SH film
Photoluminescence spectra of films made with different aggregation states DP-P3HT-SH and DP-P3HT-S-AuNPs composites film were shown in Fig. 6a and b. The PL spectra were recorded by excitation at the wavelength that the DP-P3HT-SH has maximum absorption. In this experiment, all the samples were excited at wavelength of 470 nm and the PL spectra range were collected from 480 nm to 900 nm. The DP-P3HT-SH films revealed similar emission peak at 661 nm with a shoulder at 716 nm and the PL peaks of the P3HT-S-AuNPs composites were obtained at 663 nm and a shoulder at 718 nm. | ||
| Fig. 6 Photoluminescence spectra of films made with different aggregation states DP-P3HT-SH (inset shows the PL spectra of pure AuNPs) (a), DP-P3HT-S-AuNPs composites films (b) and respective DP-P3HT-SH comparing with its composites P3HT-S-AuNPs (c–e). | ||
Fig. 6c–e showed the photoluminescence spectra of DP-P3HT-SH comparing with its P3HT-S-AuNPs composites, respectively. The PL intensity of DP-P3HT-SH has reduction upon addition of AuNPs, such as 23% for GDP-P3HT-SH, 11% for LDP-P3HT-SH and 21% for EDP-P3HT-SH. The PL intensity reduction reasons above all are morphological disorder and charge transfer reaction between the polymer P3HT-SH and inorganic nanoparticles AuNPs.15b The GDP-P3HT-SH film showed more quenching than the films made with other aggregation states after addition of AuNPs. The results revealed that the charge transfer reaction between GDP-P3HT-SH and AuNPs was the most efficient among the films made with three aggregation states, whereas the LDP-P3HT-S-AuNPs has the least charge transfer efficient. The different amount of PL quenching (23%, 11% and 21%) namely charge transfer efficient was mainly due to the different morphological structure. It could be concluded that the combination of DP-P3HT-SH and gold nanoparticle through Au–S band would increase charge transfer efficient with improving electron transfer process, resulting in increase of the amount of PL quenching.12a
Fig. 7 exhibited the PL spectra (λex = 470 nm) of three different aggregation states DP-P3HT-SH solution, DP-P3HT-S-AuNPs hybrid particles solution and respective DP-P3HT-SH comparing with its composites DP-P3HT-S-AuNPs. All the PL spectra were collected from 480 nm to 900 nm and two peaks could be observed in DP-P3HT-SH solution at about 658 nm and 720 nm which is similar to DP-P3HT-SH films. The PL spectra of composites DP-P3HT-S-AuNPs solution in Fig. 7b has the similar range and peaks compared to the films in Fig. 6b. The PL intensity quenching rate shown in Fig. 7c–e is about 18% for GDP-P3HT-SH, 10% for LDP-P3HT-SH and 9% for EDP-P3HT-SH, it could indicate the existence of charge transfer between electron donors and acceptor material. In addition, it exhibited that GDP-P3HT-SH solution has more efficient PL quenching than the other polymer. The result indicated that the materials used in solid film would improve PL quenching.
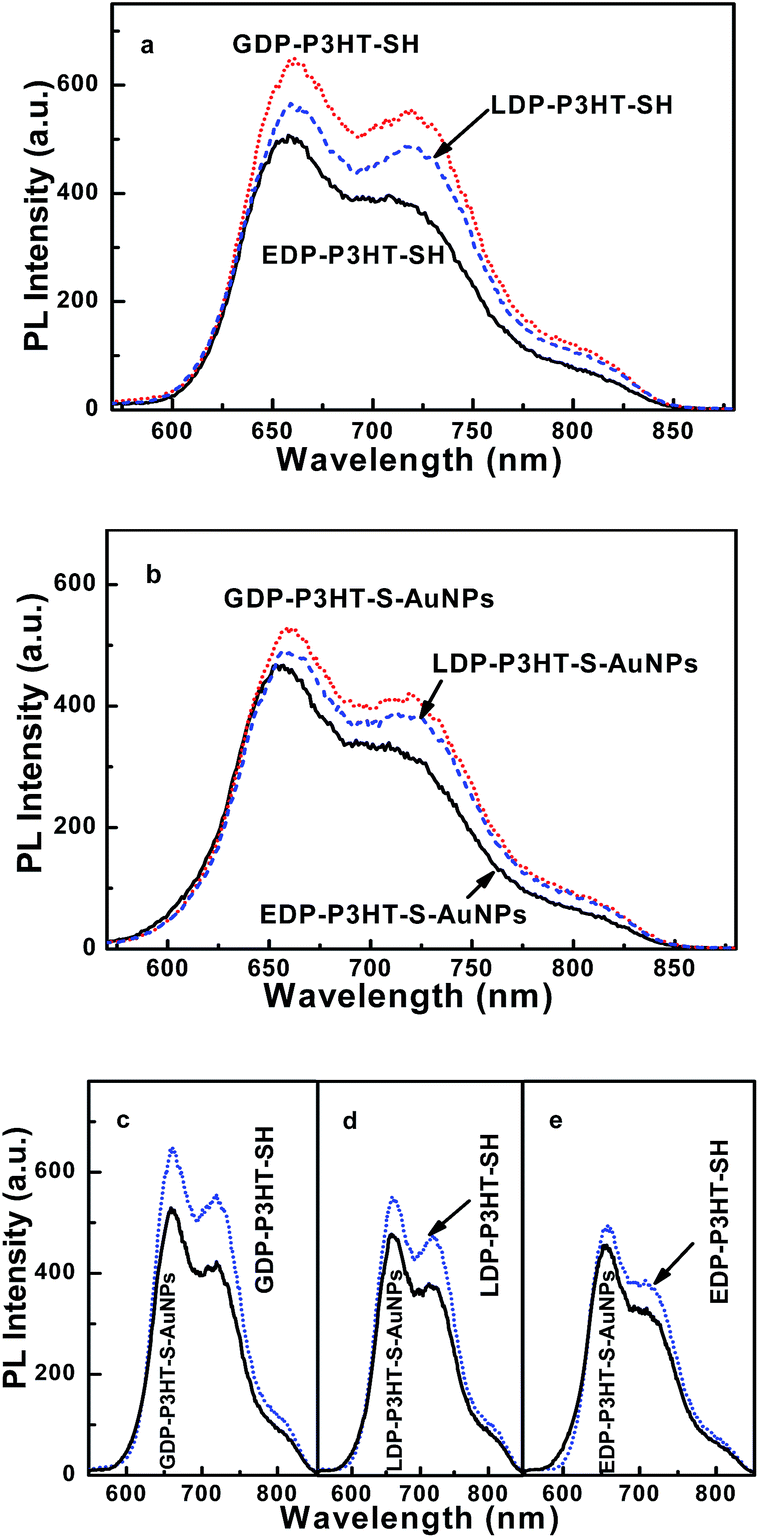 | ||
| Fig. 7 Photoluminescence spectra of different aggregation states DP-P3HT-SH solution (a), DP-P3HT-S-AuNPs hybrid particles solution (b) and respective DP-P3HT-SH comparing with its composites DP-P3HT-S-AuNPs (c–e). Conductivity of different aggregation states DP-P3HT-SH and DP-P3HT-S-AuNPs films. | ||
Fig. 8a showed the relationship between the conductivity and thickness of DP-P3HT-SH films. It can be observed that a monotonic increase trend of conductivity with increased film thickness for different aggregation state diblock copolymer DP-P3HT-SH. For GDP-P3HT-SH film, the conductivity increased from 0.5 × 10−4 S cm−1 to 1.9 × 10−4 S cm−1 with the film thickness varying from 0.8 μm to 2.4 μm. Both EDP-P3HT-SH film and LDP-P3HT-SH film exhibited similar variation trend with the range from 0.2 × 10−4 S cm−1 to 1.5 × 10−4 S cm−1 and 0.1 × 10−4 S cm−1 to 1.1 × 10−4 S cm−1, respectively.
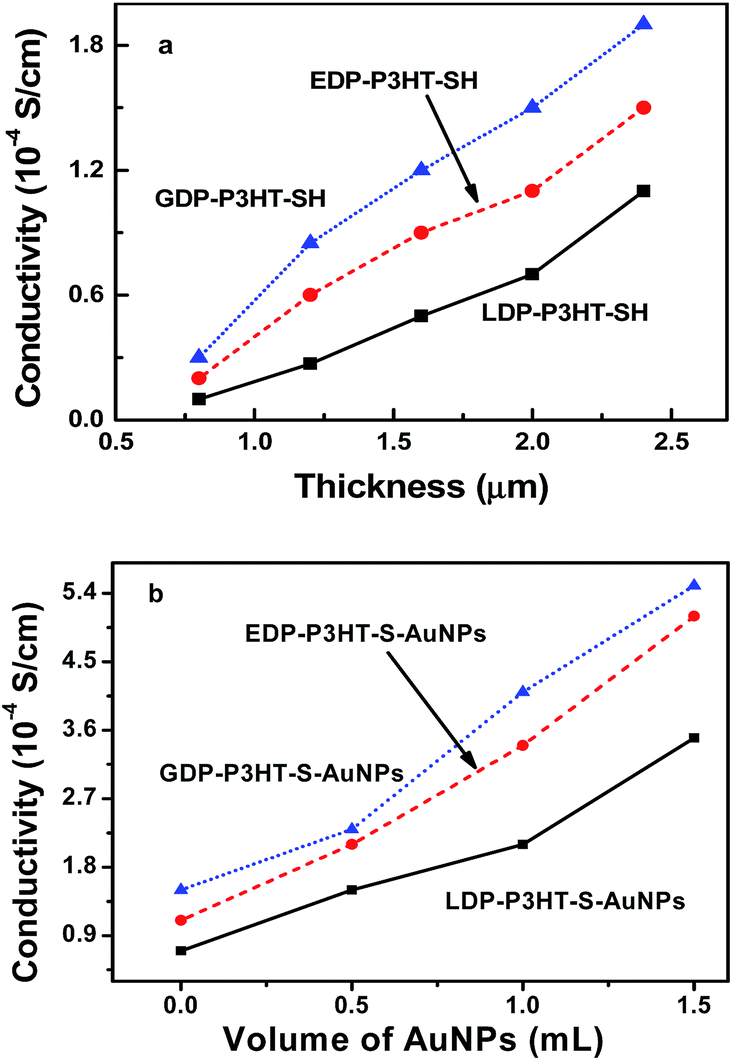 | ||
| Fig. 8 Conductivity of DP-P3HT-SH films at different film thickness (a) and DP-P3HT-S-AuNPs hybrid particles films with different volume of AuNPs (b). | ||
Although the relationship between the conductivity of P3HT and film thickness has been reported,30 there is less report on the relationship between conductivity of different aggregation states P3HT with film thickness according to the best of what we have learnt. The conductivity variation at the same thickness was attributed to the polymer aggregation state. For GDP-P3HT-SH film, each layer arranged closely and had large contact area which resulted in the best conductivity property among these three aggregation states polymer; however the LDP-P3HT-SH had large rough surface, so it presented the worst conductivity.
Fig. 8b showed the relationship between the conductivity of DP-P3HT-S-AuNPs hybrid nanoparticles films with the volume of AuNPs. The conductivity increased obviously with the addition of AuNPs. The film thickness of 2 μm was kept by drop-casting technique.25 It clearly exhibited that the conductivity of GDP-P3HT-S-AuNPs composites increased from 1.0 × 10−4 S cm−1 to 5.5 × 10−4 S cm−1, the conductivity of EDP-P3HT-S-AuNPs changed from 0.8 × 10−4 S cm−1 to 5.1 × 10−4 S cm−1, and the conductivity of LDP-P3HT-S-AuNPs varied from 0.5 × 10−4 S cm−1 to 3.5 × 10−4 S cm−1 with the increasing volume of AuNPs from 0 to 1.5 ml. Substantially the addition of gold nanoparticles enhanced the conductivity of pure DP-P3HT-SH. Gold nanoparticles were fixed on the surface of DP-P3HT-SH through thiols group to form the chemical bonds Au–S and it resulted that the gold nanoparticles distributed in polymer uniformly. In the meantime, the combination of polymer and gold nanoparticles could increase the contact area between interphase and contact ability which enhanced the ability of electron conduction. Therefore, the conductivity of composites increased with the addition of AuNPs.
In addition, the GDP-P3HT-S-AuNPs composites had the strongest conductivity among DP-P3HT-S-AuNPs composites made by three aggregation states and the LDP-P3HT-S-AuNPs composite had the lowest conductivity. The conductivity of these P3HT-S-AuNPs composites differed with each other mainly attributed to the different aggregation states of polymer. The gold nanoparticles distributed on the surface of GDP-P3HT-SH and integrated closely which led to GDP-P3HT-S-AuNPs composites having the best conductivity property. The EDP-P3HT-S-AuNPs composites had the similar binding morphology. However the LDP-P3HT-S-AuNPs composites were distinct from them because gold nanoparticles had entered into its interior which could be observed through TEM shown in Fig. 3D, and it resulted the worst conductivity property of LDP-P3HT-S-AuNPs among these three aggregation states.
Conclusion
DP-P3HT-SH had been synthesized by the simple solution and different aggregation states DP-P3HT-SH and their respective composites of DP-P3HT-S-AuNPs hybrid nanostructures had been prepared. The gold nanoparticles were attached on the surface of DP-P3HT-SH polymer. The DP-P3HT-S-AuNPs composites exhibited enhanced and wide range of absorption compared to the same aggregate state of DP-P3HT-SH. However, the photoluminescence intensity of DP-P3HT-S-AuNPs was lower than that of DP-P3HT-SH due to the addition of gold nanoparticles. The GDP-P3HT-S-AuNPs had the most amount of PL quenching about 23%; while the LDP-P3HT-S-AuNPs had the least amount of PL quenching about 11%. The charge transfer between GDP-P3HT-SH and AuNPs was the most efficient among the films made with three aggregation states, whereas, the LDP-P3HT-SH had the least charge transfer efficient.The relationships between conductivity and the AuNPs content in DP-P3HT-S-AuNPs composites, and thickness of DP-P3HT-SH films were systematically investigated. The conductivity of composites increased with the addition of AuNPs and GDP-P3HT-S-AuNPs composites showed the strongest conductivity among the DP-P3HT-S-AuNPs made by three aggregation states, whereas the LDP-P3HT-S-AuNPs composites had the lowest conductivity. In addition, every aggregation state DP-P3HT-SH had a monotonic increase trend of conductivity with increased film thickness.
Accordingly, aggregation state ultimately impacted the optoelectronic properties in solid state and the addition of gold nanoparticles could improve optoelectronics performance. Therefore this work could play an important role in achieving new optoelectronics material to modulate efficient optoelectronics devices by tuning different aggregation state diblock copolymer.
Acknowledgements
The authors thank A/Prof. D. Jin and A/Prof. H. Chen (Shanghai University, China) for support on the measurement of TEM and UV. This work was supported by the National Natural Science Foundation of China (Grant no. 51203088).Notes and references
- (a) A. C. Arias, F. Endicott and R. A. Street, Adv. Mater., 2006, 18, 2900–2904 CrossRef CAS; (b) G. M. Newbloom, K. M. Weigandt and D. C. Pozzo, Macromolecules, 2012, 45, 3452–3462 CrossRef CAS; (c) L. Martinez, S. Higuchi, A. J. MacLachlan, A. Stavrinadis, N. C. Miller, S. L. Diedenhofen, M. Bernechea, S. Sweetnam, J. Nelson, S. A. Haque, K. Tajima and G. Konstantatos, Nanoscale, 2014, 6, 10018–10026 RSC; (d) P. T. Wu, G. Ren, F. S. Kim, C. Li, R. Mezzenga and S. A. Jenekhe, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 614–626 CrossRef CAS; (e) J. Cui, D. E. Martínez-Tong, A. Sanz, T. A. Ezquerra, E. Rebollar and A. Nogales, Macromolecules, 2016, 49, 2709–2717 CrossRef CAS.
- (a) R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205–9216 CrossRef CAS; (b) B. A. G. Hammer, F. A. Bokel, R. C. Hayward and T. Emrick, Chem. Mater., 2011, 23, 4250–4256 CrossRef CAS; (c) D. H. Kim, Y. D. Park, Y. Jang, H. Yang, Y. H. Kim, J. I. Han, D. G. Moon, S. Park, T. Chang and C. Chang, Adv. Funct. Mater., 2005, 15, 77–82 CrossRef CAS; (d) S. Rajaram, P. B. Armstrong, B. J. Kim and J. M. Fréchet, Chem. Mater., 2009, 21, 1775–1777 CrossRef CAS; (e) K. Sivula, C. K. Luscombe, B. C. Thompson and J. M. Fréchet, J. Am. Chem. Soc., 2006, 128, 13988–13989 CrossRef CAS PubMed.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- X. Yu, K. Xiao, J. Chen, N. V. Lavrik, K. Hong, B. G. Sumpter and D. B. Geohegan, ACS Nano, 2011, 5, 3559–3567 CrossRef CAS PubMed.
- B. A. Hammer, M. A. Reyes-Martinez, F. A. Bokel, F. Liu, T. P. Russell, R. C. Hayward, A. L. Briseno and T. Emrick, ACS Appl. Mater. Interfaces, 2014, 6, 7705–7711 CAS.
- I. Botiz, A. B. Martinson and S. B. Darling, Langmuir, 2010, 26(11), 8756–8761 CrossRef CAS PubMed.
- I. Botiz and S. B. Darling, Mater. Today, 2010, 13(5), 42–51 CrossRef CAS.
- (a) Y.-H. Lin, S. B. Darling, M. P. Nikiforov, J. Strzalka and R. Verduzco, Macromolecules, 2012, 45(16), 6571–6579 CrossRef CAS; (b) R. Verduzco, I. Botiz, D. L. Pickel, S. M. Kilbey, K. Hong, E. Dimasi and S. B. Darling, Macromolecules, 2011, 44(3), 530–539 CrossRef CAS.
- (a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed; (b) A. Babel and S. A. Jenekhe, Synth. Met., 2005, 148, 169–173 CrossRef CAS.
- S. L. Fronk, C.-K. Mai, M. Ford, R. P. Noland and G. C. Bazan, Macromolecules, 2015, 48, 6224–6232 CrossRef CAS.
- L. Bu, T. J. Dawson and R. C. Hayward, ACS Nano, 2015, 9(2), 1878–1885 CrossRef CAS PubMed.
- (a) B. Jana, S. Bhattacharyya and A. Patra, Phys. Chem. Chem. Phys., 2015, 17, 15392–15399 RSC; (b) G. Jiang, A. S. Susha, A. A. Lutich, F. D. Stefani, J. Feldmann and A. L. Rogach, ACS Nano, 2009, 3, 4127–4131 CrossRef CAS PubMed; (c) A. Wight and M. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS PubMed; (d) R. Shenhar and V. M. Rotello, Acc. Chem. Res., 2003, 36, 549–561 CrossRef CAS PubMed; (e) A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS PubMed; (f) V. Ruiz, P. G. Nicholson, S. Jollands, P. A. Thomas, J. V. Macpherson and P. R. Unwin, J. Phys. Chem. B, 2005, 109, 19335–19344 CrossRef CAS PubMed; (g) S. Banerjee, S. C. Pillai, P. Falaras, K. E. O'shea, J. A. Byrne and D. D. Dionysiou, J. Phys. Chem. Lett., 2014, 5, 2543–2554 CrossRef CAS PubMed; (h) L. Martinez, S. Higuchi, A. J. MacLachlan, A. Stavrinadis, N. C. Miller, S. L. Diedenhofen, M. Bernechea, S. Sweetnam, J. Nelson, S. A. Haque, K. Tajima and G. Konstantatos, Nanoscale, 2014, 6, 10018–10026 RSC.
- (a) G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS; (b) J. Jo, S. I. Na, S. S. Kim, T. W. Lee, Y. Chung, S. J. Kang, D. Vak and D. Y. Kim, Adv. Funct. Mater., 2009, 19, 2398–2406 CrossRef CAS; (c) G. Zardalidis, A. Pipertzis, G. Mountrichas, S. Pispas, M. Mezger and G. Floudas, Macromolecules, 2016, 49, 2679–2687 CrossRef CAS.
- M. D. Heinemann, K. von Maydell, F. Zutz, J. Kolny-Olesiak, H. Borchert, I. Riedel and J. Parisi, Adv. Funct. Mater., 2009, 19, 3788–3795 CrossRef CAS; N. Radychev, I. Lokteva, F. Witt, J. Kolny-Olesiak, H. Borchert and J. Parisi, J. Phys. Chem. C, 2011, 115, 14111–14122 Search PubMed.
- (a) W. J. Beek, M. M. Wienk and R. A. Janssen, Adv. Mater., 2004, 16, 1009–1013 CrossRef CAS; (b) F. Li, Y. H. Du and Y. W. Chen, Thin Solid Films, 2012, 526, 120–126 CrossRef CAS; (c) W. J. Beek, M. M. Wienk and R. A. Janssen, Adv. Funct. Mater., 2006, 16, 1112–1116 CrossRef CAS.
- W.-P. Liao, Y.-H. Su, Y.-K. Huang, C.-S. Yeh, L.-W. Huang and J.-J. Wu, P3HT-based nanoarchitectural fano solar cells, ACS Appl. Mater. Interfaces, 2014, 6, 17993–18000 CAS.
- Y. Li, P. Lu and M. Jiang, et al., Femtosecond Time-Resolved Fluorescence Study of TiO2-Coated ZnO Nanorods/P3HT Photovoltaic Films, J. Phys. Chem. C, 2012, 116, 25248–25256 CAS.
- M. Achermann, J. Phys. Chem. Lett., 2010, 1, 2837–2843 CrossRef CAS.
- C. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297–6301 CrossRef CAS PubMed.
- S. Miyanishi, K. Tajima and K. Hashimoto, Macromolecules, 2009, 42, 1610–1618 CrossRef CAS.
- P. Bäuerle, F. Würthner and S. Heid, Angew. Chem., Int. Ed., 1990, 29, 419–420 CrossRef.
- L. Zhai, R. L. Pilston, K. L. Zaiger, K. K. Stokes and R. D. McCullough, Macromolecules, 2003, 36, 61–64 CrossRef CAS.
- G. Frens, Nature, 1973, 241, 20–22 CAS.
- B. Li, C. Ni and C. Y. Li, Macromolecules, 2008, 41(7), 2754 CrossRef CAS.
- J. Y. Chen, Z. B. Chen, Y. P. Qu, G. H. Lu, F. Ye, S. S. Wang, H. Y. Lv and X. N. Yang, RSC Adv., 2015, 5, 1777–1784 RSC.
- X. Xiao, Z. Hu, Z. Wang and T. He, J. Phys. Chem. B, 2009, 113, 14604–14610 CrossRef CAS PubMed.
- Y. Li, L. Song and Y. Qiao, RSC Adv., 2014, 4(101), 57611–57614 RSC; M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 7, 801–802 RSC.
- M. K. Mayeda, W.-F. Kuan, W.-S. Young, J. A. Lauterbach and T. H. Epps III, Chem. Mater., 2012, 24, 2627–2634 CrossRef CAS.
- R. Ramani and S. Alam, Polymer, 2013, 54, 6785–6792 CrossRef CAS.
- W. Li, K. H. Hendriks, W. Roelofs, Y. Kim, M. M. Wienk and R. A. Janssen, Adv. Mater., 2013, 25, 3182–3186 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the synthetic products. See DOI: 10.1039/c6ra19895c |
| This journal is © The Royal Society of Chemistry 2016 |