
Interaction of organodialkoxysilanolates with carbon dioxide†
Abstract
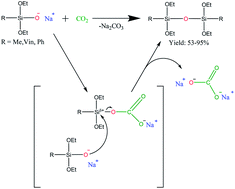
A series of organo(alkoxy)disiloxanes was obtained by the reaction of carbon dioxide with sodium alkoxy(organo)silanolates under high pressure as well as by bubbling CO2 through the reaction mixture. It has been suggested that the reaction involves the intermediate formation of carbonate derivatives of sodium alkoxy(organo)silanolates.
 Please wait while we load your content...
Please wait while we load your content...

