
Simultaneous and direct analysis of multiple types of organic contaminants in water based on a MOF decorated with a suitable quantity of Au nanoparticles, using SALDI-TOF MS†
Abstract
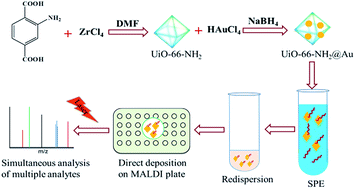
For the first time, surface-assisted laser desorption/ionization time-of-flight mass spectrometry (SALDI-TOF MS) combined with direct solid-phase extraction was used to simultaneously and rapidly analyze multiple types of organic contaminants in water samples. The technique employed gold nanoparticles (Au NPs) loaded onto the metal–organic framework (MOF) UiO-66-NH2. A suitable quantity of Au NPs endows excellent matrix functionality to the nanocomposite with only weak Au cluster peak interference.
 Please wait while we load your content...
Please wait while we load your content...

